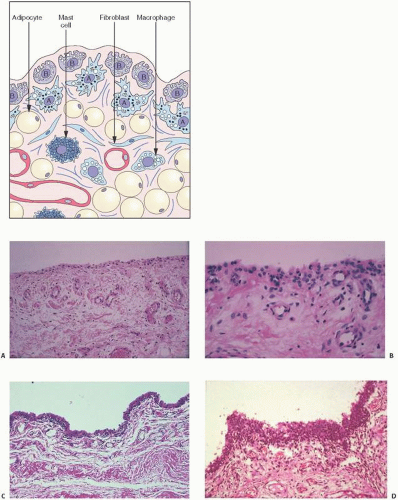
Hemosiderotic Synovitis
In hemorrhages that occur within the synovium, a highly vascularized tissue, red blood cells eventually disintegrate with phagocytosis occurring by histiocytes. Eventually, the hemoglobin is broken down and processed into hemosiderotic. Hemosiderotic characteristically is seen as a brown-pigmented granular substance in cells of the synovium. Hemosiderotic may occur as minute granules or accumulate into globules 25 µm in diameter. The intracellular deposition of hemosiderotic is observed mostly in macrophages or histiocytes, but also may be seen engulfed in various trauma-associated conditions by the hypertrophied synovial lining cells. In joints subjected to hemorrhage, hemosiderotic may be noted intracellularly or extracellularly. Cells within different compartments or layers of the synovium may be affected, ranging from the synovial lining cells to the subsynovial proliferating connective tissue cells. The gross appearance of the joint with chronic hemarthrosis is that of a rusty pigmentation, although more acute bleeding may be demonstrable as a blackish green discoloration.
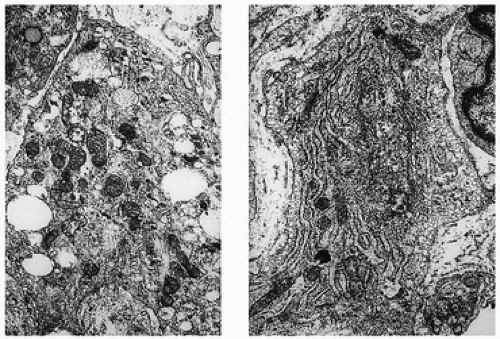
The response of the human joint to bleeding is denoted clinically by the formation of a hyperplastic vascular synovium within a few days. Examination of the tissue reveals a proliferation of the synovial cells and other subsynovial lining cell connective tissue elements, including often inflammatory cells. Under electron microscope, iron-containing, electron-dense particles that are membrane bound, called siderosomes, are noted within the synovial cells and subsynovial macrophages.
Clinically, Hemosiderotic synovitis may be due to a wide range of conditions, most notably trauma, particularly chronic trauma leading to chronic hemarthrosis. It may also be caused by the use of oral anticoagulant therapies, as a result of breakdown of synovial hemangiomas, or as a secondary phenomenon in conditions such as rheumatoid arthritis, PVNS, scurvy, and sickle cell anemia. The radiographic appearances may lead to joint space narrowing and may be confused with other ailments destructive to joints. Although most patients with chronic hemarthrosis or with an episode of hemarthrosis recover without any significant sequelae, the potential for joint destruction is present in any patient with bleeding into the joint.
With bleeding into the joint, two pathways may lead to damage. In one mechanism, the red blood cells may break down, causing macrophage activation. The ensuing inflammation may lead to destructive changes in and of itself, as seen in rheumatoid arthritis. Intracellular hemosiderotic precipitates the release of leukocyte-derived and synovium-derived chondrolytic enzymes (
17). In excessive cases, the iron deposition in organs such as the meniscus may be severe enough to cause mechanical dysfunction and degenerative changes. In a second pathway, bleeding may lead directly to synovial proliferation, the release of an uncontrollable cascade of destructive substances such as proteases.
Hemophilia
Hemophilia A, the most common hereditary coagulation disorder, affects 20 live male infants per 100,000. It is caused by the deficiency, absence, or malfunction of coagulation factor VIII (
18). In normal patients, a very low concentration of factor VIII (0.2 mg/mL plasma) is sufficient for adequate coagulation. Bleeding leading to clinically perceptible illness requires a reduction of at least 75 percent. The level of factor deficiency determines the severity of the disease: severe (factor VIII or IX baseline activity <1 percent), moderate (residual activity 1 to 5 percent), or mild (residual activity >5 percent).
Hemophilia A is a clinically heterogeneous disorder, ranging from mild (1 to 4 percent deficiency) to severe disease. Patients with mild or moderate disease may not be recognized unless a significant traumatic event precipitates abnormal bleeding. Excessive bleeding during surgery may be the first clue. Therapy centers around replacement of factor VIII (
19,
20). Traditionally done with shotgun frozen plasma, cryoprecipitate became available in the 1960s. More recently, purified concentrates have been used. In general, 1U of factor VIII increases plasma activity by 0.024 U/mL. Because 0.3 U/mL is usually needed to treat a mild episode of bleeding, clinical goals should strive for more than this amount. The development of inhibitory antibodies to the factor replacement products remains a clinical challenge (
19). Therapy with recombinant concentrates have eliminated the risk of transmission of pathogens such as hepatitis and HIV viruses.
Clinically, abnormal hemorrhaging is key, especially into joints. Most commonly involved, in order of frequency, are the knees and elbows, followed by ankles, shoulders, and hips. Not usually evident in infancy, childhood signs include mild discomfort and limitation of joint motion. Pain and swelling follow. Numerous damaging microhemarthroses have transpired before clinical suspicion is initially aroused, so that the diagnosis is unfortunately delayed.
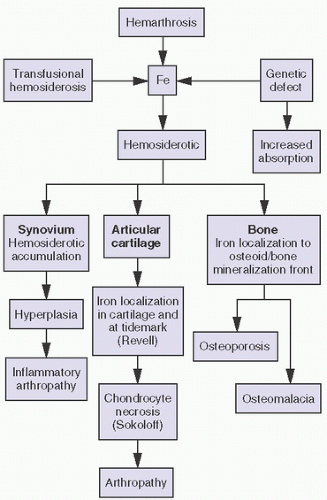
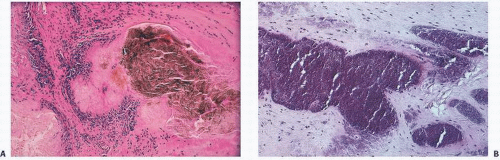
The pathology in the joint centers around iron-induced synovial inflammation (
Fig 15.11), with both clinical and experimental observations documenting the damaging effects of hemosiderotic synovitis.
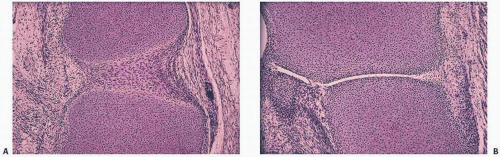
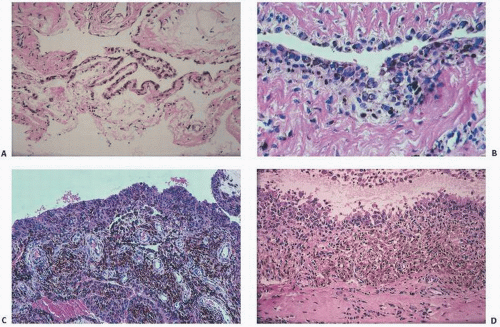
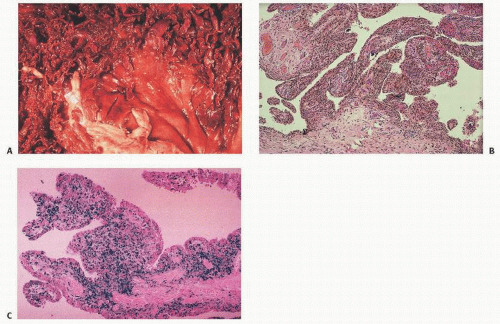
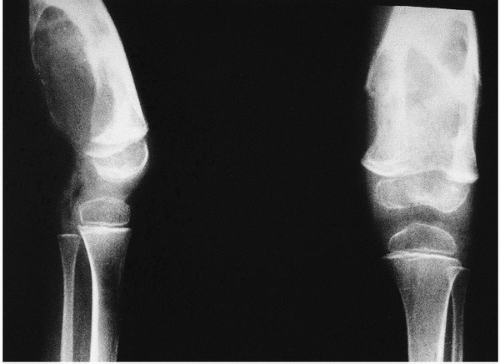
The adverse effect on synovium in patients with chronic hemorrhage has been studied most extensively in the joint changes of hemophilic arthropathy of the knee (
Fig. 15.12). In tissue removed from hemophiliacs, the synovium shows marked villous hypertrophy with extensive hypertrophy and hyperplasia of the synovial lining cells and the subsynovial connective tissue components with abundant intracytoplasmic hemosiderotic granule accumulation (
Fig. 15.11). In tissue cultures of synovium in hemophilia, pigment-laden fibroblast cells have been shown to proliferate, and explants of the synovial cells have been shown to secrete large amounts of latent collagenase and neutral proteinases, establishing the destructive potential of the synovitis in hemophilia. Synovium incited by hemophilia produces enzymes and may do so without coincident inflammation, the latter a significant component of the destructive arthropathy seen in rheumatoid arthritis.
Iron, a probable inducer of synovial inflammation, induces the expression of various oncogenes and proliferation of synovial fibroblasts. Hemosiderotic-laden tissue also leads to increased levels of proinflammatory cytokines such as interleukin 6, interleukin 1, and tissue necrosis factor alpha (
21).
Iron accumulation may lead directly to chondrocyte destruction. In experiments aimed at developing a model for PVNS, a condition secondary to expanding nodules of proliferative fibroblasts and histiocytes, blood was experimentally injected into various animal models. The resultant proliferative synovitis was characterized by hyperplasia of fibroblasts, lipid-laden macrophages and giant cells, and extensive hemosiderotic accumulation similar to that seen in reactive Hemosiderotic synovitis or hemophilic arthropathy. The experimental bleeding model does not resemble PVNS.
Although less visible, iron is known to accumulate in cartilage as well. Extensive hemosiderotic deposition leading to grossly visivble brown menisci has been observed clinically. In studying hemophilic joints, iron has been localized to superficial chondrocytes, suggesting chondrocytic phagocytic uptake triggering a degradative enzyme release similar to that described in synovial cells. Iron has been localized histochemically to the tidemark in lupus and in hemochromatosis. Hemophilia A and hemophilia B are inherited X-linked deficiencies of factor VIII and factor X.
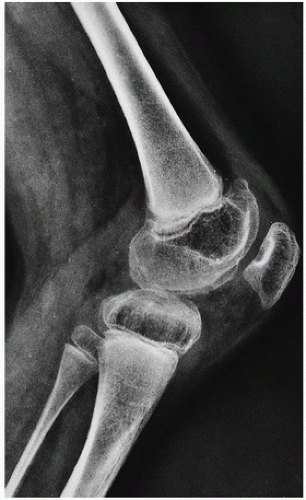
Roentgenographically, narrowing of joint spaces, loss of articular cartilage, cystic remodeling of bone (
Fig. 12.8), and hemophilic pseudotumors characterize the illness (
Fig. 15.13). However, scrupulous adherence to maintenance of factor VIII levels to prevent spontaneous hemorrhage has been associated with significantly decreased morbidity and has diminished, but not eliminated, the radiographic progression of disease.
Involvement of the musculoskeletal system is one of the major complications of hemophilia. The knee is the most commonly involved joint, followed by the elbow, ankle, shoulder, and hip.
Hemophiliac arthropathy in the small joints of the hands and feet is rare, with few series of cases described in the literature. In decreasing order of frequency, the following are the imaging findings in hemophilic arthropathy: synovial hyperplasia, erosions, subchondral cysts, cartilage loss, and “pseudotumors.”
The radiographic presentation of hemophilic arthropathy can be divided into stages for didactic purposes, but in real practice, there is frequent overlap between the different phases of the disease. Stage I is acute bleeding in the joint. Distension of the articular capsule is easily seen as a distended suprapatellar bursa in the case of the knee, or displacement of the humeral fat pads in the case of the elbow. Distension of the ankle joint capsule is usually easy to detect in the lateral projection. Bleeding into the shoulder or hip joints is difficult to determine in plain radiographs, but can be assessed by MRI and, to a lesser degree, also by computed tomography (CT).
Stage II is characterized by osteopenia in the subchondral surfaces of the affected joint. Overgrowth of the epiphyses is usually not noticeable. Joint contractures, such as fixed flexion of the knee, result in apparent enlargement of the ends of the bone on anteroposterior radiographs because of the increased distance between the knee and the x-ray cassette. This false enlargement is excluded if the lateral view is also evaluated. Enlargement of the ends of the bone is regarded to be secondary to hypervascularity, present in some cases of hemophilia. The deposition of hemosiderotic in the capsule of a joint renders it slightly more dense than the surrounding soft tissues. This is most easily detected in the elbow and ankle, but it can also be noticed in the knee.
Changes in bone contour are noted in stage III. There is squaring of the patella, widening of the intercondylar notch, and subchondral cyst formation. Care should be exercised not to confuse a tunnel view of the knee (an anteroposterior view of the knee with flexion contracture is a tunnel view) with an intercondylar notch that is really widened. The subchondral cysts tend to be larger in hemophilia than in other inflammatory arthritides.
Erosion and destructive changes in the articular cartilage result in narrowing of the joint space. The changes in the articular cartilage are secondary to intra-articular liberation of digestive enzymes, as well as to prevention of the normal nourishment by synovial fluid, which cannot diffuse into cartilage coated by blood. These abnormalities are seen in stage IV.
The final stage, stage V, is defined by extensive bone destruction with marked deformity and rigidity of the joint.
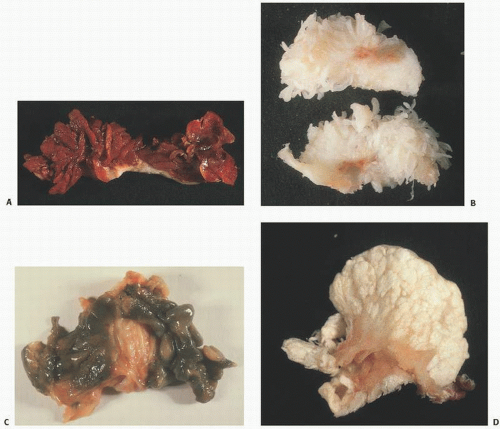
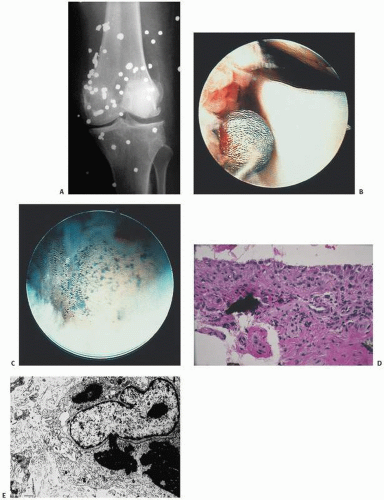
The extra-articular manifestations of musculoskeletal hemophilia include muscle hemorrhage and pseudotumors. Hemorrhage in muscles can lead to neurovascular compromise. Such hemorrhage is most common in the gastrocnemius muscle and in the volar muscles of the forearm, where it can lead to a compartment-type syndrome. Bleeding in the iliacus muscle can result in femoral nerve palsy. MRI and CT are helpful in the diagnosis of episodes of bleeding into muscles.
Pseudotumors are the result of the development of an encapsulated hematoma that slowly expands and may involve bone, the periosteum, or the soft tissue (
22). This is a rare complication in which the hemorrhagic blood fails to be resorbed, accumulates, and slowly erodes the adjacent bones. It is usually an extremely painful condition and occurs in fewer than 1 percent of patients. Pseudotumors in adults are most common in the thigh, pelvis, and retroperitoneum, and most often in the vastus lateralis, soleus, or iliopsoas muscles.
In children, they can occur in the hands and feet. Ideally, pseudotumors should be surgically removed as they can continue to grow even after long dormant periods. However, surgical removal is not without complications including death, infection, fistulization, and pathologic fractures (
23).
Surgical joint reconstruction in hemophiliacs has historically been necessary to preserve function. Although the mechanical survival of total knee replacement is quite good, the prevalence of infection as a complication is high, usually caused by
Staphylococcus epidermidis (
21).
Intra-articular, intrabursal, and soft tissue bleeding in hemophilia may result in painless masses clinically that roentgenographically mimic a tumor. These masses consist of spongy coagula of partially clotted blood encapsulated by thick, fibrous membranes. Complications of these so-called hemophilic pseudotumors include muscle and bone damage, infection, and neuropathies. Surgical removal is not without danger (
22). They occur in 1 to 2 percent of hemophiliacs, mostly in the lower extremity and pelvis. Bleeding in the vicinity of the periosteum has been implicated in the peculiar juxtaosseous changes, which have included cyst-like bone changes and large soft tissue masses eroding bone. MRI findings depend on the stage of bleeding and associated effusion, erosions, cysts, cartilage damage, and degree of iron deposition in the tissue (
24).
Before 1970, hemophilia A was still associated with significant severe disability and even death at a young age. However, the median life expectancy has increased significantly throughout the 20th century from 11.4 years to 68 years in carefully followed populations (
25).
Nonetheless, the transfusion of blood products, including factor VIII concentrates, has led to one of the well-recognized modern complications of hemophilia-transfusion-related acquired immunodeficiency syndrome (AIDS). Life expectancy has now reversed after considerable gains. Serologically detectable antibodies to human immunodeficiency virus (HIV) developed in large numbers of hemophiliacs beginning around 1979. Approximately two-thirds of HIV-positive hemophiliacs have eventually died of AIDS.
Therapy more recently has been with factor VIII concentrates exposed to vigorous virus-killing heat treatment or solvent cleaning. Genetically engineered products are now available and are being tested for complications. Expense is a significant consideration.
In treating chronic hemophilic arthropathy, standard measures include factor replacement, physiotherapy, and operative synovectomy by conventional arthrotomy or arthroscopy. Nonoperative techniques, including the destruction of synovial tissue by intra-articular injection of radioactive agents such as colloidal 32P chromic phosphate, oxytetracycline chlorhydrate (Emicine), Yttrium-90 (for knees), or Rhenium-186 (for elbows and ankles) have been used (
26).
In treating patients with HIV-positive hemophilia, the risk to the surgeon is minimal if proper precautions are taken (
27).
Hemophilia B is due to deficiency of coagulation factor IX. Also known as “Christmas disease” after a 10-year-old patient named Stephen Christmas, hemophilia B has important royal associations. Queen Victoria was its most famous carrier, and Czar Nicholas II’s son Alexei Romanov, one of its most famous patients.
Recent advances in adenovirus-associated virus vector-mediated gene transfer offer great promise in treating and reducing the cost of the disease (
28).
Iron Overload and Hemochromatosis
The most common iron overload disorders (IOD) are hemochromatosis and β-thalassemia, disorders that are typically clinically insidious. They cause progressive tissue damage including significant effects on the musculoskeletal system. Categories of iron overload and iron overload-related disease have been defined.
Screening laboratory tests include measurements of the serum ferritin level and transferrin saturation, with ferritin levels above 200 ng/mL (449 pmol/L) in women or 300 ng/mL (674 pmol/L) in men who have no signs of inflammatory disease, and transferrin saturation above 45 percent in women or above 50 percent in men warranting additional testing.
Four major cell types determine iron content and distribution (
29):
Duodenal enterocytes by affecting dietary iron absorption;
Erythroid precursors by affecting iron utilization;
Reticuloendothelial macrophages by affecting iron storage and recycling; and
Hepatocytes by affecting iron storage and endocrine regulation.
Iron released from enterocytes (and macrophages) binds to plasma iron transport protein transferrin. Transferrin transport of iron to the hepatocyte and macrophage can be stored in those cells as ferritin. Erythroid precursors are the major site of iron utilization. Hepatocytes regulate the production of the hormone hepcidin, which downregulates the release of iron via ferroportin.
IODs can be categorized according to whether there is a defect in the hepcidin-ferroportin axis (“hemochromatosis”), impaired iron transport, or ineffective erythropoiesis (e.g., “thalassemia”).
The two major options to treat iron overload are phlebotomy for hemochromatosis and iron chelators (such as deferoxamine, deferasirox, or deferiprone) for β-thalassemia in which anemia coexists with iron overload.
The hepcidin-activating pathway has recently emerged as a target for treatments aimed at modulating iron status (
30).
Much current knowledge about the effect of iron on tissue comes from studies of hemochromatosis, the systemic disorder in which iron deposition is associated with tissue dysfunction. In general, iron overload has been studied in clinical situations in which there has been hyperplastic refractory anemia, excessive blood transfusions for underlying hematopoietic abnormalities (thalassemia), and hereditary forms of hemochromatosis. In the United States and northern Europe, hereditary hemochromatosis is a common cause of iron overload; 15 percent of the population exhibits the gene in its heterozygous state. The abnormal gene, located on the short arm of chromosome 6, is closely linked to the HLAA locus.
In hereditary hemochromatosis, there is an increased absorption of dietary iron, resulting in excess iron deposition in parenchymal tissues of endocrine organs, liver, and heart; death often results from heart failure or chronic liver disease. In other IODs, the cause is ineffective erythropoiesis as seen in β-thalassemia and sideroblastic or aplastic anemia, for which the treatment may include repeated transfusions. IODs also may be caused by chronic liver disease, and in certain populations, such as within sub-Saharan Africa, hemochromatosis has been attributed to increased amounts of dietary iron intake, such as from large amounts of iron in beer brewed in steel drums. Individuals at greatest risk probably have a genetic predisposition (
29).
The deposition of iron in bone, particularly at the osteoblast-osteoid interface or at the mineralization front or as hemosiderosis within the hematopoietic cells of the marrow, is a relatively common sequela to primary and transfusional hemosiderosis. Of import, iron directly inhibits osteoblast differentiation. Although the primary organs involved are the heart and the liver with eventual development of cirrhosis and hepatocellular carcinoma and heart failure, endocrine organs are particularly involved. Joint pain in the clinical presentation of arthritis has been noted in 11 percent in some studies, but its prevalence is as high as 72 percent. Bone pain is a less frequent cause for presentation (
31) (
Fig. 15.14,
Table 15.4).
First recognized in 1964 as a feature of hemochromatosis, arthropathy is now generally believed to be a common clinical symptom. In hemochromatosis, the joint changes may mimic DJD. Less than 50 percent of patients seem to have clinically osteoarticular problems. The metacarpophalangeal joints and interphalangeal joints are most frequently reported. Wrists, elbows, shoulders, hips, and knees are less frequently affected. Iron may inhibit the pyrophosphatase activity in cartilage tissue, leading to the precipitation of calcium pyrophosphate crystals, an association between the two that is well recognized.
Bone changes in iron overload have been described in hemochromatosis as well as the iron-overloaded states of chronic renal dialysis. Osteoporosis is common, particularly if concomitant hypogonadism was present.
The osteopathy seen in iron overload has been associated with various concurrent metabolic abnormalities including hypogonadism, hyperparathyroidism, diabetes, and both vitamin C and vitamin D deficiency. Hypogonadism is a significant contributing factor to osteoporosis in this population, with both bone resorption parameters increased (osteoclast surfaces, osteoclast numbers, and urinary hydroxyproline levels), and bone formation parameters decreased. In hemochromatosis, iron may also infiltrate and adversely affect the parathyroid gland, with previous studies showing concurrent elevated parathyroid hormone.
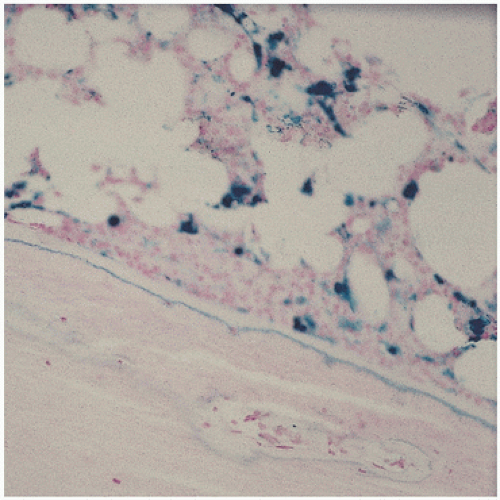
Iron overload associated with hemodialysis can lead to a wide range of musculoskeletal problems. Proximal muscular weakness is well known and may be due to a number of problems including concomitant endocrinopathies, phosphate depletion, and osteomalacia. Although iron deposition at the mineralization front has been shown in numerous conditions including thalassemia, its histochemical detection in undecalcified bone biopsies may or may not be associated with osteomalacia (
Fig. 15.14). In fact, as mentioned in hemochromatosis, the most common finding is osteoporosis, even with iron accumulation at the mineralization front. Tidemark iron accumulation has been demonstrated in articular cartilage, as well as in patients with hemochromatosis.
In the osteomalacia seen in association with renal disease, the disorder is usually the result of the toxic effects of aluminum, and, in fact, aluminum deposition at the mineralization front is now well established in the literature as a common cause of osteomalacia in this population. However, the presence of iron, obvious in dialysis patients by the significant hemosiderosis that complicates many cases, can also be the cause of osteomalacia (
Fig. 4.4). Although iron may be the sole cause in rare cases of osteomalacia, in many cases it may also be deposited along with aluminum, causing peculiar staining properties at the mineralization front.
Although intestinal absorption of iron is normal in patients on hemodialysis, 1,25-vitamin D may stimulate iron absorption. However, it is more likely that iron deposition causing osteomalacia in the dialysis population is due to transfusion overload.