of bone biopsy specimens from large series of patients who have long-standing end-stage renal disease with or without hemodialysis has revealed a broader range of changes, which include secondary hyperparathyroidism, osteomalacia, or a mixture of osteomalacia and secondary hyperparathyroidism. Histomorphometric studies in which tetracycline labeling is used have also revealed a type of ROD characterized by sparse cellular activity and low bone turnover (aplastic bone) (6) (Table 4.1). In addition, tissue samples from these patients may or may not have detectable aluminum and/or iron deposition at the mineralization fronts. Rare circumstances of iron at the mineralization front causing osteomalacia have been described (7). There are also patients with an increasing range of amyloid-related syndromes resulting from the deposition of β2-microglobulin (8). An additional unusual tissue change, which is most evident on x-ray films, is osteosclerosis, a condition characterized by increased bone deposition in tissue despite obvious identifiable osteoclastic resorption.
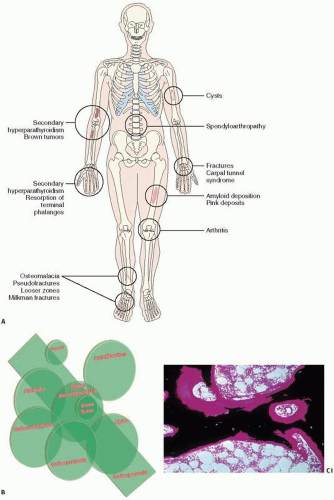 FIGURE 4.1. Renal Osteodystrophy at a glance. (A,B) Clinical and roentgenographic abnormalities in renal-related bone disease. (C) Osteomalacia showing increased volume and surface of osteoid. (C1) (Continued) |
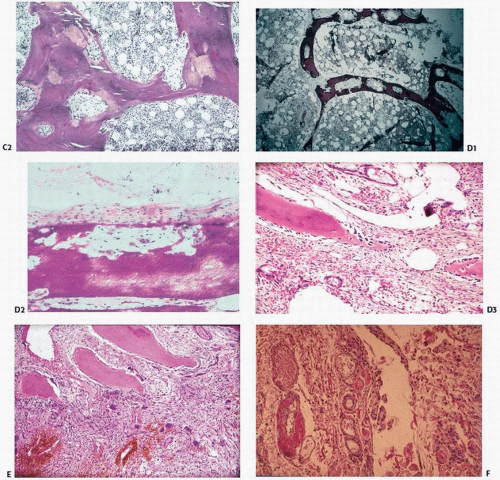 FIGURE 4.1. (Continued) Von Kossa stain. (C2) H&E stain. (D1) Hyperparathyroidism showing increased bone resorption as evidenced by lytic holes in the trabecular bone. (D2) Subperiosteal osteoclastic bone resorption, and (D3) high remodeling bone with fibrotic marrow. (E) Brown tumor showing giant cells clustering around areas of hemorrhage. (F) Amyloid deposition (pink) deposition in vessel walls. |
TABLE 4.1 Histomorphometric and Histologic Classification of Renal Osteodystrophy | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||
phosphate retention,
↓ calcitriol levels,
↓ serum ionized calcium,
↓ numbers of vitamin D receptors and ↓ numbers of calcium sensors in the parathyroid gland,
skeletal resistance to the calcemic action of PTH due to ↓ density of PTH receptors on osteoblasts; ↑ serum osteoprotegerin levels.
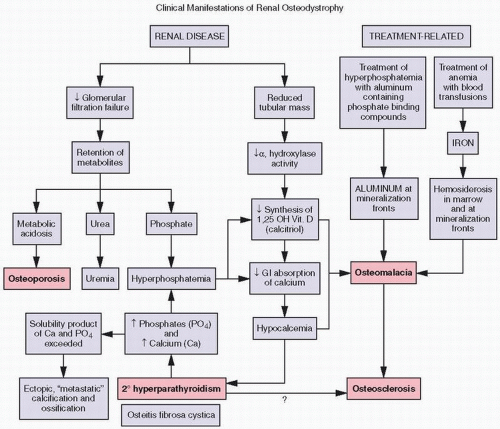 FIGURE 4.2. Biochemical (purple boxes) and osseous histologic (pink boxes) abnormalities in renal osteodystrophy. |
study of the uptake and activity of tetracycline at the bone surface, reveal at least five recognizable types of bone remodeling disease in ROD: (a) secondary hyperparathyroidism, (b) osteomalacia, (c) mixed uremic dystrophy (secondary hyperparathyroidism and osteomalacia), (d) adynamic or low-turnover disease, and (e) osteosclerosis (11). Stains for aluminum, iron, and amyloid should be performed, as all these may contribute to pathology.
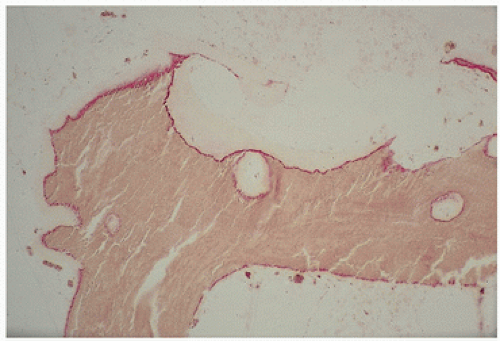 FIGURE 4.3. Aluminum (red) osteomalacia in renal-related bone disease develops at the osteoid-bone interface (“mineralization front”) and is directly associated with osteoid accumulation. |
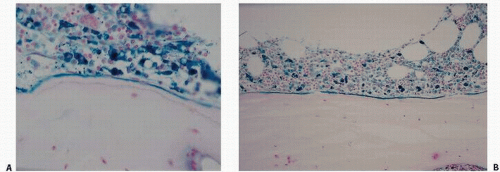 FIGURE 4.4. Iron (blue)-related bone disease in renal osteodystrophy includes marrow hemosiderosis with its potential impact on bone-lining cells as well as accumulation either along marrow-osteoid sites (A) or at the mineralization front (B), seen also in aluminum-induced osteomalacia. |
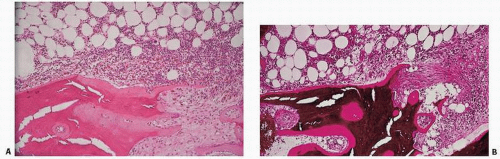 FIGURE 4.5. Osteitis fibrosa is the paratrabecular and tunneling fibrosis of the marrow seen in association with renal disease in patients who have biochemical evidence of secondary hyperparathyroidism. Fibrosis occurs at the surface and is associated with hematopoietic hypercellularity adjacent to it. (A) Hematoxylin-eosin (H&E) stain. (B) von Kossa stain, undecalcified sections. |
multiple, “brown tumors” represent the disorder von Recklinghausen described as osteitis fibrosa cystica, a rare observation in modern times due to early diagnosis and treatment of hyperparathyroidism.
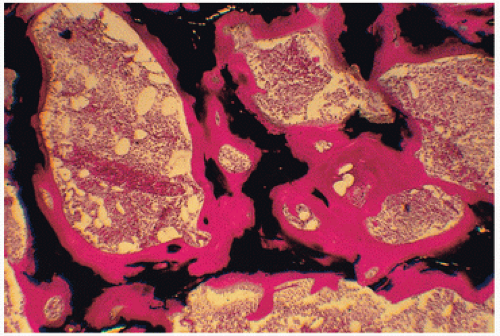 FIGURE 4.6. Osteomalacic bone in renal disease demonstrates marked accumulation of osteoid (red), which in normal states constitutes only 2 percent of the volume of bone and covers only 20 percent of its surface. (Undecalcified bone, von Kossa stain.) |
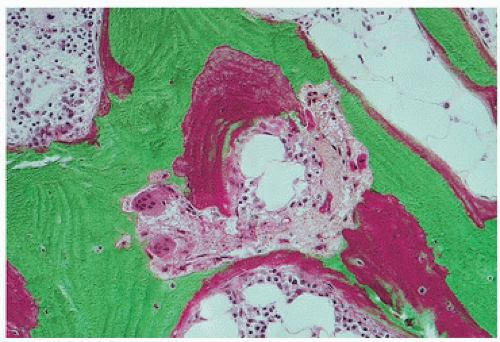 FIGURE 4.7. Osteoid accumulation and osteoclastic tunneling resorption in mixed uremic bone disease. Increased tunneling resorption substantiates secondary hyperparathyroidism. Osteoid accumulation when shown by tetracycline labeling to be indistinct and smudged with decreased mineralization rates is consistent with osteomalacia. |
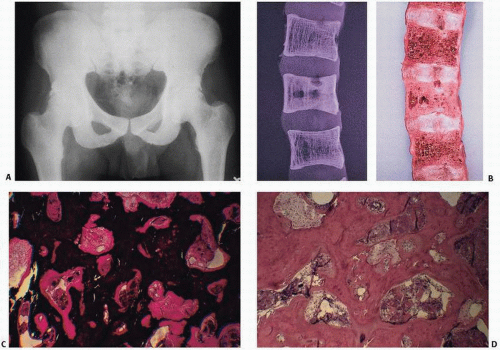 FIGURE 4.8. Osteosclerotic variant of renal osteodystrophy is characterized by diffuse or localized radiodense bone (A). Gross examination of the spine (B) and x-ray films of gross specimens may reveal diffuse obliteration of marrow space by thickened cortical and trabecular bone. Undecalcified biopsy specimens reveal abundant mineralized bone encroaching on the marrow space (C) (von Kossa stain). Foci of osteoclastic resorption document the ability of these patients to resorb bone, but suggest a dysfunctioning remodeling event (D) (H&E stain). (Continued) |
increase in systemic calcium load, will limit soft tissue calcification and potentially reduce cardiovascular risk.
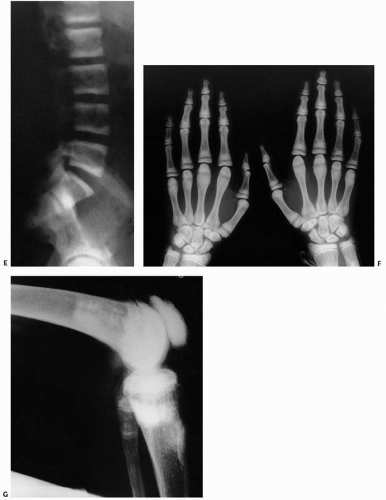 FIGURE 4.8. (Continued) Other classic roentgenographic features include alternating bands of lucency and sclerosis (“rugger jersey” spine), as seen in a lateral radiograph (E). In a frontal view of both hands and wrists, sclerosis is seen in the metaphyses and epiphyses of most of the short tubular bones, carpal bones, and distal ends of the radii and ulnae. There is expansion of the metaphyses of the metacarpals and mild widening of the growth plates, most apparent in the proximal phalanges of both thumbs (F). In a lateral view of the knee, areas of sclerosis are seen in the metaphyses and epiphyses of the femur and knee as well as in the patella (G). |
appearance of amorphous, eosinophilic deposits under light microscopy after hematoxylin and eosin (H&E) staining,
bright green birefringence under polarized light after staining with the dye Congo red,
regular fibrillary structure observed with electron microscopy,
β-pleated sheet structure demonstrated with x-ray diffraction.
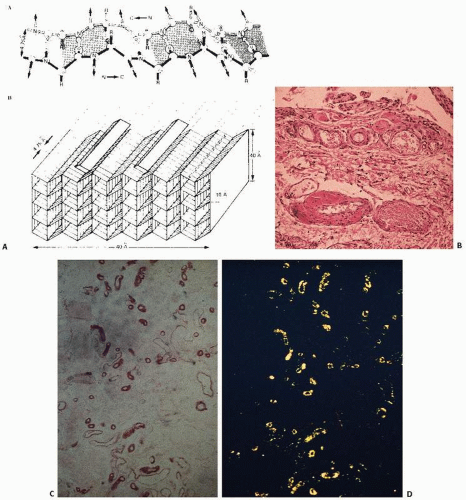 FIGURE 4.9. Amyloid composite. Fibrillary protein with a biochemical β-pleated structure (A) appears pink on routine tissue stain and is usually perivascular in location (B). With Congo red staining (synovial biopsy), amyloid tissue is pinkish red (C), and it is characterized by an apple green birefringence on polarized light microscopy (D). (Continued) |
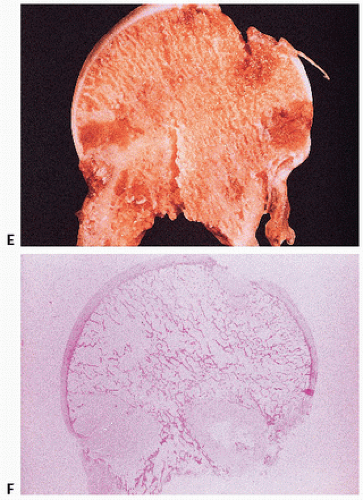 FIGURE 4.9. (Continued) Gross (E) and photomicroscopic (F) appearance of amyloid of the femoral head. (After Sack GH Jr, Dumars KW, Gummerson KS, et al. Three forms of dominant amyloid neuropathy. Johns Hopkins Med J. 1981;149:239-247.) |
ocular and renal pathology are seen. Treatment is directed at reducing the amount of methionine (a dietary precursor of cystine) and cystine in the diet or using drugs that can reduce intracellular cystine levels. When possible, renal transplantation should be performed to bypass the genetically determined defect in the intracellular environment.
TABLE 4.2 Nomenclature and Classification of Human Amyloid and Amyloidosis | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
TABLE 4.3 Syndromes and Clinical Manifestations of β2-Microglobulin Amyloid Deposition | ||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||
within membrane-limited organelles, having the appearance and enzyme markers of lysosomes, that is, containing acid phosphatase. The specific localization of crystals within lysosomes has suggested a classification of cystinosis as a lysosomal storage disease.
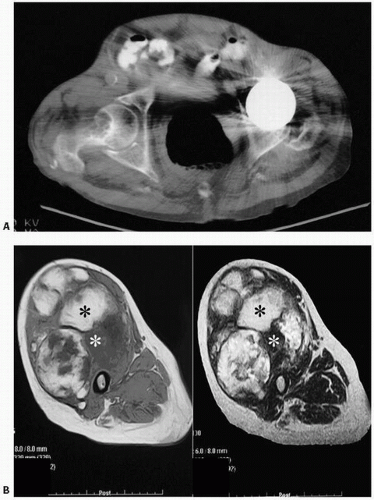 FIGURE 4.10. Amyloid. (A) Computed tomographic scan of the hip: well-defined lucency in the right femoral neck of a patient undergoing long-term renal dialysis. The opposite femoral head has been replaced because of osteonecrosis. The radiographic finding in amyloidosis is usually of one or more well-defined lucencies surrounded by minimal sclerosis. The lesions tend to occur in the ends of long bones. Metaphyseal and diaphyseal lesions can be found at times. The most common sites include the proximal femora, long bones at the knee, long bones of the upper extremity, and spine. (B) Axial T1-weighted and T2-weighted spin echo MR images of the right thigh show the mass involving the entire anterior compartment as well as the tensor fasciae latae. The mass is again noted to demonstrate both hypointense and hyperintense areas. |
TABLE 4.4 Some Distinguishing Characteristics of the Three Major Types of Cystinosis | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
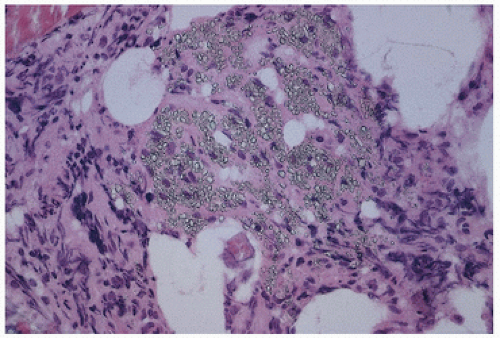 FIGURE 4.11. Cystine crystals in tissue. |
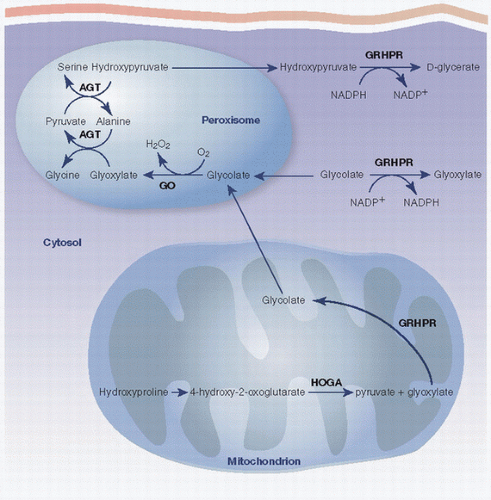 FIGURE 4.12. Oxalate metabolism. |
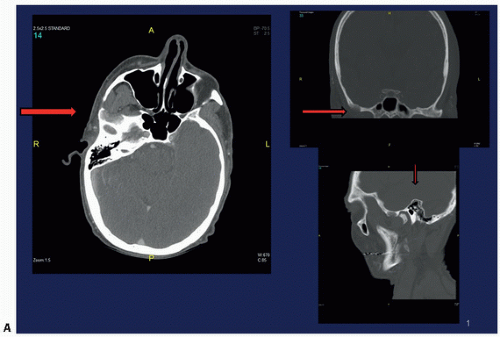 FIGURE 4.13. Oxalosis. Lytic lesion in the right infratemporal fossa (A). (Continued) |
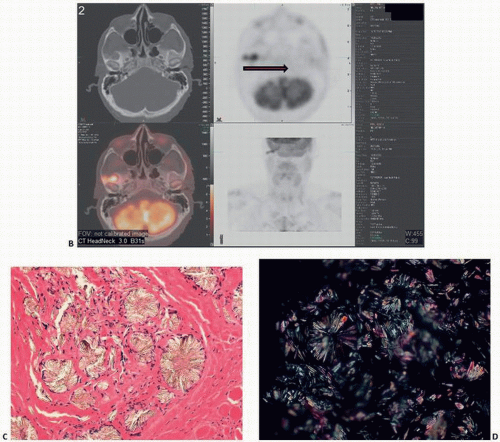 FIGURE 4.13. (Continued) Pet scan shows bilobed focus of hypermetabolic activity (standardized uptake values initially 4.4 and 3.0, and 8.1 and 5.8 on delayed imaging) (B). Histology of the crystals with flower and star shapes (C). polarized light microscopy (D). |
 FIGURE 4.14. Grotesque facial features are due to enlarged frontal skull bone and maxilla. (After Quinten Metsys [1465/66-1530], The National Gallery, London.) |
TABLE 4.5 Symptoms of Paget’s Disease | ||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||
to fracture, and, in fact, fracture may be the presenting symptom. Other associated changes include calcific periarthritis and uremia, neurologic symptoms including deafness, vascular steal syndromes, platybasia (flattening of the base of the skull), and basilar invagination. Immobilization can lead to hypercalciuria, hypercalcemia, and nephrolithiasis. Roentgenographically, the changes are variable, depending on the stage and site of involvement (Fig. 4.16). Purely lytic lesions in Paget’s disease have been described, particularly in the skull, so-called osteitis circumscripta. In the initial stages of Paget’s disease in long bones, a lytic wedge advances along the cortex of the bone. Biopsies at this stage may show predominantly osteoclastic bone resorption mimicking hyperparathyroidism. Eventually, the resorptive phase progresses to one of intense bone remodeling, including osteoblastic bone formation. The resultant deterioration of the normal architecture of the bone leads to roentgenographically detectable coarsening of cortical bone, lack of demarcation between cortical and trabecular bone, and other such findings. The bone scan in this phase is intensely hot (Fig. 4.17). In many patients, Paget’s disease after a prolonged period of time enters a dormant phase, with little detectable osteoblast or osteoclast activity. In this stage, the disorder can be diagnosed by profound thickening and irregular cement lines throughout the involved bones. Fracture after biopsy of osteolytic Paget’s disease (82,83) and acute osteolysis after surgery potentially predisposing the patient to pathologic fracture (84) have been described.
TABLE 4.6 Pagetic Involvement of Different Bonesa | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||
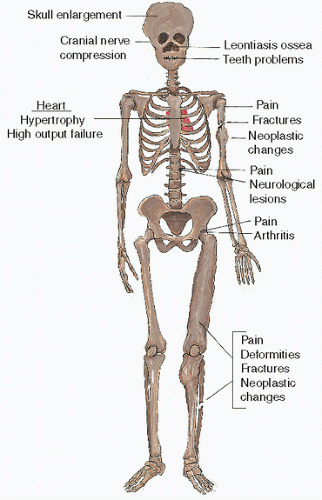 FIGURE 4.15. Clinical symptoms of Paget’s disease. |
advancing flame or the tip of an arrow). The deformity in the contour of the bone is the consequence of fractures (complete or incomplete). Fractures in bones with Paget’s disease heal in an irregular fashion, resulting in deformities (osteitis deformans). The deformity in the contour of the end of the bone can at times result in nonunion, narrowing of the adjacent joint space, and osteophyte formation. This form of degenerative joint disease has been labeled Paget’s arthritis, and it can occur when only one bone is involved, even though it is more common when both bones surrounding a joint are affected.
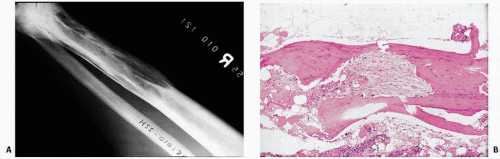 FIGURE 4.16. Roentgenographic (A) and histologic (B) changes of Paget’s disease. Areas of sclerosis and lucency extend from the distal end of the bone into its shaft. The proximal end of the area of Paget’s has a pointed edge. |
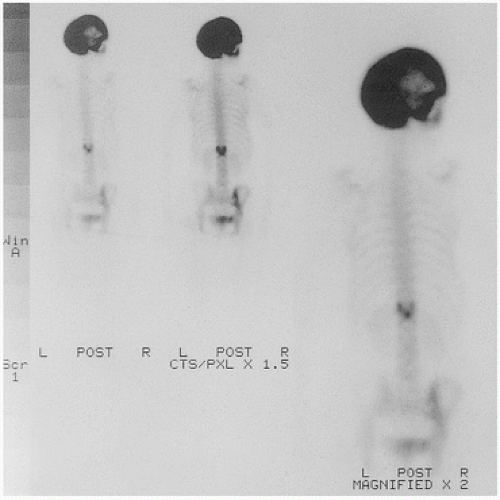 FIGURE 4.17. Hot skull (bone scan). |
formation and bone resorption. Bone trabeculae become both thick and thin. Cortical bone remodeling is also irregular, with loss of the distinction between cortical and trabecular bone. Osteoclasts are increased in number, large, and markedly multinucleated.
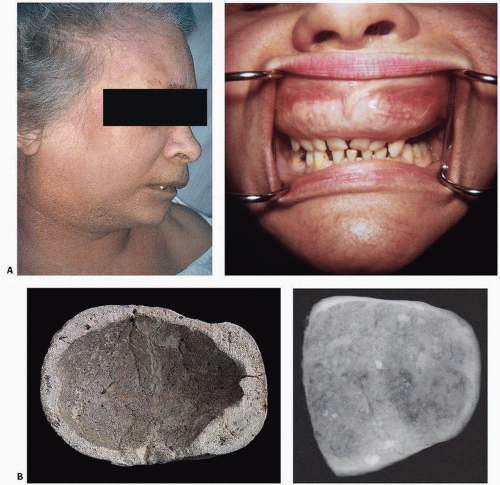 FIGURE 4.18. Composite of gross bone changes. (A) Paget’s disease involving the maxilla appears as a bulging upper lip (left), and oral examination reveals an enlarged distorted maxilla with loose teeth (right). (Courtesy of Ernest Baden, M.D., D.D.S.) (B) Calvarium showing irregular thickening of the skull, loss of diploic architecture, and a granular pumicelike appearance (left). Specimen x-ray film (right) shows irregular density with alternating densities and lucencies. (C) Proximal end of femur (left) and specimen x-ray slice of hip and acetabulum (right) reveal loss of normal cortical and cancellous architecture with replacement by coarsened, thickened bundles of bone. Grossly, bone may appear irregular, coarse, and pinkish, in contrast to the normal smooth, ivory appearance of cortical bone. (D) Vertebrae involved by Paget’s disease reveal sclerosis, typically more marked at the vertebral end plate, giving rise to a “picture frame” contour (left). External contour and internal architecture of bone are greatly distorted. Gross specimen and specimen x-ray film (right). (E) Undecalcified transcortical iliac bone biopsy specimen stained with von Kossa stain showing loss of demarcation between cortical and trabecular bone distorting the internal architecture of the ilium (left). Higher power (right) reveals irregular surfaces, increased surface osteoid, marrow fibrosis, and hypervascularity. |
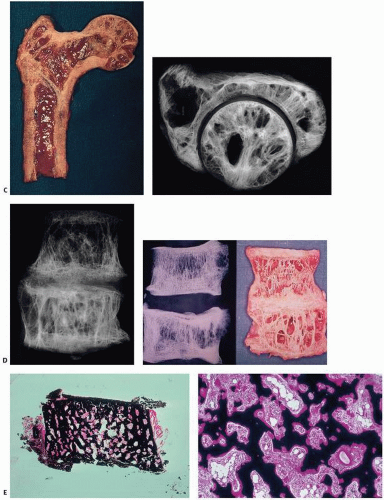 FIGURE 4.18. Continued |
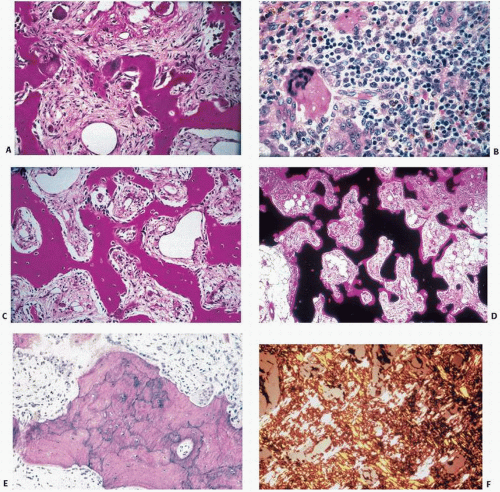 FIGURE 4.19. Paget’s disease at a glance. • Abnormal osteoclasts Large and multinucleated (A) Numerous osteoclasts (B) • Increased osteoblast activity Increased numbers of osteoblasts (C) • Increased bone remodeling. Increased activation frequency (D) • Woven bone Irregular cement lines (E) Irregular collagen orientation (F) (Continued) |
lines (Fig. 4.22) in now “burned-out” dense and (relatively) inactive tissue.
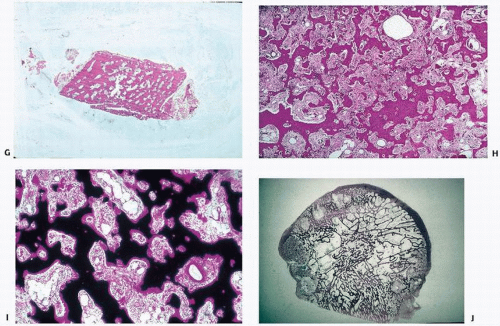 FIGURE 4.19. (Continued) • Poor cortical/cancellous bone demarcation (G) • Stromal fibrosis (H) • Increased vascularity (I) • Focal sharp demarcation from normal bone (J) |
TABLE 4.7 Iliac Trabecular Bone Morphology and Histomorphometry Data in Normal Bone and Case Examples of Idiopathic Osteoporosis, Osteomalacia, Secondary Hyperparathyroidism, and Paget’s Disease | |||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||
inclusions have been shown in osteoclasts in Paget’s disease by Rebel et al. (87) and Reddy et al. (88). However, osteoclast inclusions are by no means specific and have been shown in giant cells in giant-cell tumors (89), pyknodysostosis (90), osteopetrosis (91), familial expansile osteolysis (92), and in macrophage-like cells in primary oxalosis (93).
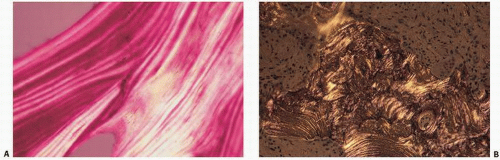 FIGURE 4.20. Polarized light histopathology of normal bone (A) and pagetic bone (B). Normal bone reveals the predictable lamellar, sheet-like orientation of collagen. In Paget’s disease, this is distorted, resulting in less refractile, interrupted wisps of collagen, “woven bone.” |
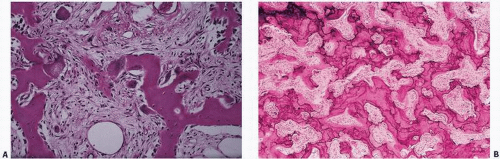 FIGURE 4.21. Active Paget’s disease with marked fibrosis and osteoclast activity (A) and pronounced irregular cement lines (B). |
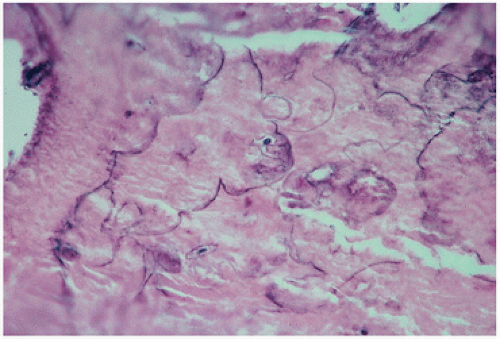 FIGURE 4.22. Inactive, “end-stage” or burned-out Paget’s disease. Only the curliform cement lines remain as a marker of the disease. |
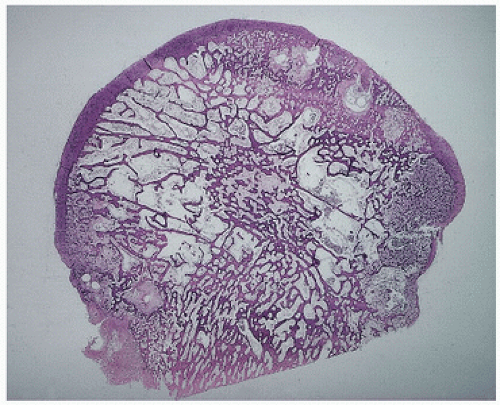 FIGURE 4.23. Coronal section through a femoral head with degenerative joint disease and Paget’s disease. The circumscribed dense areas represent pagetic foci. Intervening bone was nondiagnostic. |
Osteoclasts are irregularly shaped with multiple extensions and much infolding of cytoplasmic membrane. The infolding presumptively indicates abnormally increased surface activity and motility of these cells.
The presence of calcified fragments within the cytoplasm under the ruffled border shows that the cells may actually phagocytose whole pieces of bone, which is a highly abnormal form of bone resorption.
The presence of vesicular mitochondria suggests a high turnover rate for osteoclasts.
Nuclei are highly polymorphic and frequently contain many nucleoli. Nuclear inclusions are present in several nuclei per osteoclast and are filament-like structures. Some are microcylindric in shape. They are usually grouped together in parallel bundles and sometimes packed in paracrystalline arrays.
Cytoplasmic inclusions can be filament-like structures, some of which are organized into bundles, like those found in the nucleus. No paracrystalline arrays, however, are usually found in the cytoplasm. The cytoplasm may also contain envacuolated glycogen.
In the fibrous tissue surrounding the osteoclasts in Paget’s disease, cells similar to mononuclear osteoprogenitor cells are often found. These are often massed together and contain no nuclear inclusions.
TABLE 4.8 Paget’s Tumors | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||
 FIGURE 4.24. Skeletal distribution of Paget’s sarcoma. |
late stage at presentation compared with conventional osteosarcoma
higher percentage of metastatic disease at presentation
more rapid metastatic spread
higher risks in the use of chemotherapy due to higher age and poorer health of patients
risks associated with Paget’s disease-related high-out left ventricular heart failure
more axial location of Paget’s sarcoma compared with conventional osteosarcoma
harder detection of the tumors due to patient age-related mental frailty
desmoplastic fibroma (105). The most extensively evaluated benign lesion is the so-called Avellino tumor (Fig. 4.26).
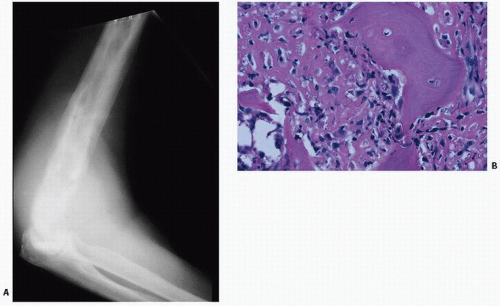 FIGURE 4.25. Paget’s sarcoma. Roentgenogram of Paget’s sarcoma of the humerus with superimposed destructive lytic lesion (A), proven to be an anaplastic sarcoma on biopsy (B). |
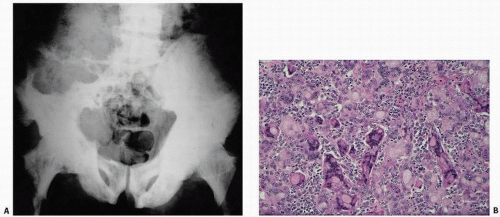 FIGURE 4.26. Giant-cell lesion in Paget’s disease. An 86-year-old white male immigrant from Avellino, Italy, with worsening pain in both extremities and weakness and progressive inability to ambulate. Physical examination revealed a large, left-sided, soft, immobile, painless mass predominantly involving the left buttock. Pelvic x-rays reveal diffuse pagetic changes of the pelvis, lumbar spine, sacrum, both pubic rami, and both proximal femora, and thickening of the iliopectineal line, all of which are classic for Paget’s disease of bone (A). A large lytic lesion was observed eroding the left iliac wing, L4, L5, and the sacrum. Computed tomogram was remarkable for a left-sided soft tissue mass invading the iliac crest, lumbar spine, and spinal canal. Pathology revealed multinucleated giant cells of various sizes and shapes, often with peculiar clear inclusions (B).
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|