Fig. 5.1
Biosynthesis of 3ʹ-phosphorylated phosphatidylinositol. a. An example of a typical form of PtdIns found within the plasma membrane of cells. A range of kinases can phosphorylate the 4, 5, and 3 carbon atomes within the inositol ring structure. b. The normal sequence of phosphorylation of PtdIns and the enzymes responsible for each step. PI3 kinase is normally the rate liiting enzyme in this sequence. (Reproduced from (O’Neill 2008a))
The formation of polyphosphorylated PIP3 within the membrane creates a docking site for a wide range of important signalling proteins (Fig. 5.2). This in turn allows for the aggregation of effector molecules into functional transducsomes. There is a large diversity of possible transducsomes that could be generated in response to PIP3 formation. Many of the effectors typically activated by PI3K activate pro-survival and anti-apoptotic pathways (Datta et al. 1999; Downward 2004).
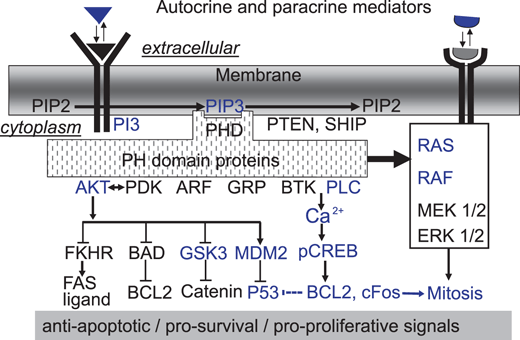
Fig. 5.2
Possible signalling pathways activated by embryotropic ligands. Extracellular tropic ligands are implicated in PI3K-mediated survival signalling. There is also some evidence implicating activation of the RAS signalling pathway. Ligand-mediated activation of PI3K results in the formation of PIP3 which act as a docking site for a host of PH-domain containing proteins. To date only AKT and PLC have been investigated in detail and found to be implicated in survival signalling in the early mouse embryo. The activation of AKT can in turn induce changed activity of a host of survival mediators, and of these the MDM2/TRP53 pathway has been best studied. The PLC pathway causes increased intracellular calcium levels which can activate the CREB transcription factor, which in turn generates a pro-proliferative/anti-apoptotic transcriptome. The pathways and mediators shown in blue are those for which direct experimental support exists in the embryo. Those in black are known from other cellular models but are yet to be validated in the early embryo
The PI3K survival pathway is present within cells of the early embryo and is activated by autocrine trophic ligands (Li et al. 2007b; Lu et al. 2004). The essential role of this pathway is illustrated by the lethality of early embryos in which the P110 beta (Pik3cb−/−) component of PI3K is genetically deleted (Bi et al. 2002). The embryotropins Paf (Lu et al. 2004), insulin, IGF1 (Navarrete Santos et al. 2008) and TGFα (Kawamura et al. 2005) have all been implicated in the activation of the PI3K pathway in the early embryo. Pharmacological inhibition of PI3K (LY294002 (3 µM) or wortmannin (10 nM)) compromised early development of the embryo and also blocks signal transduction events as early as the zygote and 2-cell stage of development (Lu et al. 2004). The infusion of 2-cell embryo cells with an anti-PIP3 antibody blocked signal transduction events initiated by Paf, while direct infusion of PIP3 itself can mimic the action of Paf (Li et al. 2007b). Taken together these results provide a compelling case for the role of PI3K in mediating the actions of Paf and other embryotrophins.
The adverse effects of PI3K inhibitors (both wortmannin and LY294002) on preimplantation embryo development in vitro were partially ameliorated by communal culture or by the exogenous administration of Paf. Acute treatment of mouse blastocysts with LY294002 (50 μM) induced an increased incidence of apoptosis (TUNEL staining) within 10 h of treatment (Gross et al. 2005), implicating the PI3K pathway in a survival response by the embryo. Immunolocalization revealed the presence and membrane localisation of PI3K throughout the preimplantation stage of development (Riley et al. 2005).
Pharmacological analysis indicated that a range of trophic ligands act in an overlapping (and potentially redundant) manner to activate PI3K signalling (Lu et al. 2004). The rate at which PIP3 is degraded is central to the strength and duration of any cellular response to a ligand. PIP3 is inactivated by a selective phosphatase: phosphatase and tensin homolog deleted on chromosome ten (PTEN) (also known as mutated in multiple advanced cancers 1, MMAC1). PTEN reverses the action of PI3K by converting PIP3 to PIP2. Genetic deletion of PTEN results in early embryonic demise (before day 7.5 of development, but detailed analysis of the developmental defect has not been made) (Cristofano et al. 1998). The persistence of PIP3 in cells would be expected to occur in the absence of PTEN, and the lethality of Pten−/− embryos suggest that tight control of temporal responses to these signal is an important requirement of normal development.
5.5 Downstream Responses to PIP3
A current paradigm of cell biology is that all cells require the action of a range of survival factors to maintain the latency of constitutively active effectors of cell death (Raff et al. 1994). In the absence of such survival factors it is proposed that cells enter into a default process of apoptotic cell death. It is argued that this is a mechanism for providing positional information to cells during development (and during subsequent life) that facilitates tissue remodelling and helps guard against ectopic or metastatic growth of cells. Survival signalling induced by trophic ligands has been studied in many somatic cell types and is commonly transduced by the action of PIP3.
The apparent developmental autonomy of the early embryo distinguishes it from all cells at later stages of development and postnatally, where normal cells have a requirement for exogenous ligands. This autonomy seems to be explained by the survival signal actions of the autocrine embryotrophins such as Paf, via their activation of PI3K (O’Neill 2008).
The canonical survival signalling actions of PI3K occurs as a consequence of the formation of PIP3 which can act as a docking site for a range of proteins, that contain the pleckstrin homology domain (PH) (Lemmon 2007). The PH domain comprises approximately 120 amino acids and consists of two perpendicular anti-parallel beta-sheets followed by C-terminal amphipathic helix. This domain is one of the most common domains within the mammalian proteome (Lemmon 2007). Proteins known to interact with PIP3 include a range of transcription factors , kinases, cytoskeletal proteins, effectors of lipid and carbohydrate metabolism. This diversity of targets illustrates the potential of this pathway to generate a range of responses and also hints at considerable plasticity in the embryo’s responses to its environment.
An important function of PIP3 is that it allows recruitment of kinases and their substrates to facilitate phosphorylation. One important example of this form of interaction involves 3-phosphoinositide-dependent protein kinase 1 (PDK1). PDK1 is considered a master regulator of the AGC family of kinases (cAMP-dependent, cGMP-dependent and protein kinase C) (Mora et al. 2004). It also has critical roles in coordinating activation of other downstream PH-domain proteins such as RAC-alpha serine/threonine-protein kinase (AKT).
An essential role for the activation PDK1 in early development is demonstrated by genetic deletion studies. Conditional knockout of the maternal copy of Pdk1 resulted in infertility. Normal fertilisation of eggs occurred but they subsequently arrested at the 2-cell stage (Zheng et al. 2010). This arrest resulted from the failure of the normal embryonic genome activation, but without any obvious effect of progression of embryos through the G1/S phases of the cell-cycle. Loss of maternal PDK1 and the absence of expression of paternally expressed PDK1 caused a marked reduction of the overall levels of transcription in the 2-cell embryo and of the synthesis of hallmarks of embryonic genome activation, such as the TRC protein complex. The actual arrest at the 2-cell stage seems related to the restricted formation of MPF activity at the G2-M interface.
An important finding that confirms the role of PIP3 in these actions of PDK1 was that the co-deletion of maternal PTEN rescued the two-cell arrest. During culture in vitro for 3 days, from E1.5 to E4.5, all Pdk1-null embryos arrested at the 2-cell stage while most of the Pdk1:Pten double null embryos reached the four-to-eight-cell stage and then the blastocyst stage. This result infers that excessive accumulation of PIP3 can compensate for the loss of PDK1. The mechanism of this compensation has yet to be defined but one attractive possibility is that under these conditions sufficient PIP3 accumulates to result in the activation of AKT independently of PDK1.
5.6 AKT
AKT is phosphorylation by PDK1, and the resulting activated protein is an important cell survival signal (Datta et al. 1999; Downward 2004). The early embryo expresses three genes that encode AKT variants (Akt1-3) (Li et al. 2007b), and immunostaining with a pan-AKT antibody decorates each stage of early embryo development (Riley et al. 2005). Activation of AKT upon its binding to PIP3 and association with PDK1 results in phosphorylation of serine 473 of AKT. This activation state modification is detected in the early embryo. Treatment of embryos with Paf (Li et al. 2007b) or insulin (Riley et al. 2005) cause enhanced AKT activation within the early embryo. A chimeric protein that possesses the PH domain from AKT coupled with a GFP-reporter was found to associate with the membrane of two-cell embryos under conditions that favour autocrine signalling (Halet et al. 2008).
AKT is one of its best investigated targets of PI3K signalling. It possesses a PH domain and so docks with PIP3. In this way it can typically form close associations with PDK1. PIP3 can activate PDK1 which in turn phosphorylates PIP3-bound AKT. The resulting serine 473 phosphoAKT is detected in all stages of preimplantation development in the mouse (Riley et al. 2005), and exposure of 2-cell embryos to Paf in vitro caused an up-regulation in its expression (Li et al. 2007b). Acute treatment with insulin increased the level of phosphorylated AKT, as assessed by western blot analysis in the mouse (Riley et al. 2005) or rabbit (Navarrete Santos et al. 2008). In the rabbit blastocyst, insulin induced phosphorylation of AKT in the trophectoderm but not in the inner cell mass, while IGF-1 created the opposite response. This result indicates that lineage specificity of PI3K signalling may occur in the early embryo , and highlights the potential for context-specific cellular responses to a given embryotrophic ligands.
The binding of PH-domain proteins to PIP3 can facilitate signalling in a least 3 ways: (1) binding can induce conformational changes in the bound protein allowing, for instance, autophosphorylation (Standaert et al. 2001); (2) it can induced allosteric changes in the bound protein resulting in changes to its function, such as, the relief of pseudosubstrate autoinhibition (Standaert et al. 2001); and (3) binding can bring the other PH domain substrate proteins into close proximity with PH domain containing kinases, facilitating phosphorylation (Rebecchi and Scarlata 1998). It is generally considered that the interaction of PDK1 and AKT is an example of the later form of activation. Yet the compensation of PTEN loss for loss of PDK1 suggests that under conditions of increased PIP3 critical downstream effects might be activated independently of PDK1, perhaps by increased activity of the first two forms of activation. This question requires further investigation.
Both genetic and pharmacological analysis shows that AKT activation is critical for early embryo development. There are three AKT genes so conventional gene knock-out approaches are challenging. Yet, injection of mRNA coding for a constitutively active myristoylated AKT into mouse zygotes enhanced their rate of cell-cycle progression, conversely mRNAof kinase-deficient AKT (Akt1-KD) delayed the entry of zygotes into mitosis. It was further shown that AKT induced the phosphorylation of cell division cycle 25 homolog B (CDC25B), and hence induced MPF activity (Feng et al. 2007). Two structurally unrelated inhibitors of AKT activity (deguelin, 5–15nM and AKT Inhibitor, 3–10 μM) induced a dose-dependent reduction in rates of zygote development to the blastocyst stage in vitro (Li et al. 2007b). The embryopathy induced by AKT inhibition was partially reversed by the addition of exogenous Paf to culture medium. The similar inhibitory profiles of putative PI3K (Lu et al. 2004) and AKT inhibitors (Li et al. 2007b) are consistent with these agents acting within the same signalling pathway in the early embryo. Furthermore, Paf partially reversed the adverse effects of both PI3K and AKT inhibition on embryo development. As noted above, serine 473 phosphoAKT is detected in all stages of preimplantation development in the mouse (Riley et al. 2005), and exposure of 2-cell embryos to Paf in vitro caused an up-regulation in its expression (Li et al. 2007b), a result confirming the normal role of trophic ligand signalling via PDK1 in the activation of AKT-mediated cell survival.
5.7 PI3K-Dependent Calcium Signalling
Other PH-domain containing proteins have critical roles in a range of signalling pathways including the activation of transient increases in intracellular calcium concentration. Phospholipase C gamma (PLCgamma) is a PH domain protein and may be an important component of Paf-mediated calcium signalling . The two PLCgamma genes (Plcg1 & 2) (Li et al. 2007b) and immunodetectable PLCgamma are expressed in the early embryo (Li et al. 2007b; Wang et al. 2007). PLCgamma hydrolyses PIP2 to form inositol trisphosphate and diacylglycerol. Inositol trisphosphate is a critical component of ligand-mediated transient increases in the cells intracellular calcium concentration ([Ca2+]i), and its activity is required for Paf-induced [Ca2+]i transients in 2-cell embryos (Emerson et al. 2000). Selective inhibition of PLC inhibits Paf-induced calcium transients (Emerson et al. 2000). Paf also activates the influx of calcium via a dihydropyridine-sensitive calcium channel (Emerson et al. 2000; Lu et al. 2003), and such channels are known to be activated downstream of PI3K (Kamp and Chiamvimonvat 2006; Le Blanc et al. 2004). These signalling responses were dependent upon the presence of the Paf-receptor in the embryo.
The calcium transient induced by Paf was accompanied by a marked hyperpolarisation of membrane potential and this was caused by a net outward ion current (~ 280 pA). This current is composed of a 4,4′-diisothiocyanatostilbene-2,2′-disulfonate-sensitive (anion channel blocker) and tetraethylammonium chloride sensitive (K+ channel blocker) channels (Li et al. 2007b; Li et al. 2007c). Direct infusion of PIP3 (1,2-dipalmitoyl-sn-glyceryl-3-phosphatidylinositol-3,4,5-trisphosphate) into the cytoplasm of the 2-cell embryo mimicked the action of Paf in inducing the activity of this current and membrane hyperpolarization. The role of membrane hyperpolarisation in response to Paf is currently not known. Such changes in membrane potential are commonly associated with changes in membrane transport function, for example, and this role of trophic signalling warrants more extensive investigation. The calcium signal and the ion channel function of Cl– seems essential for the trophic signalling responses of Paf because the inhibition of either causes loss of their developmental viability (O’Neill 2008a, b).
The pulses of intracellular calcium that occur in the early embryo in response to Paf persist for around 5 min and the embryo then becomes desensitised for a period of around 90–120 min until they again become responsive to Paf (Emerson et al. 2000). This process of response, followed by desensitization and subsequent re-sensitization provides a mechanism for generating periodic responses to autocrine survival signalling in the early embryo. These pulses commenced at the mid-zygote stage and peaked in amplitude at the mid 2-cell stage (Emerson et al. 2000), the time when PDK1-dependent activation of embryonic genome activation and generalised increases in transcriptional activity occurred (Zheng et al. 2010).
Calcium signalling has many potential targets within cells. Detailed analysis in the early embryo shows that the Paf-induced calcium transients activate a calmodulin/calmodulin-dependent kinase induced phosphorylation and activation of the CREB transcription factor (Jin and O’Neill 2007, 2010, 2011). This activation of CREB in the embryo occurs at the time of activation of definitive transcription from the embryonic genome (Jin and O’Neill 2007, 2010).
CREB has the potential to generate a very extensive transcriptome (Impey et al. 2004; Zhang et al. 2005) and its activation at the time of definitive activation of the transcription from the zygotic genomic could conceivably have a major role in directing this transcription. The CREB transcriptome is recognised as critical to survival of a range of somatic cells and during oncogenesis (Kinjo et al. 2005; Shaywitz and Greenberg 1999; Siu and Jin 2007; Walton and Dragunow 2000). CREB is one member of a family of transcription factors . Other members of this transcription factor family include CREM and ATF1. Members of this family act as dimers to form active transcription factors. Each member of the family can either homodimerise or bind with another members of the family to form active heterodimers. Each of these dimer pairs has different regulatory inputs and is likely to generate a slightly different transcriptome. Analysis of the early embryo shows that CREB and ATF1, but not CREM, are expressed in the early mouse embryo (Jin and O’Neill 2010). In the early embryo it is likely that the CREB-ATF1 heterodimer is the most critical for development. Creb −/− ,Atf −/− double knockout embryos degenerate by the late blastocyst stage and this is due to extensive apoptotic death of cells within the embryo (Bleckmann et al. 2002). The high incidence of cell death in embryos of the double knock-out indicates that the CREB :ATF1 dimer may be the active transcription factors critical in generating the transcriptome coding for cell survival in the preimplantation embryo. This implicates an important role of trophic ligand-induced activation of CREB in the formation of this active transcription factor dimmer.
Within the very diverse transcriptome potentially generated by the CREB family of transcription factors are two canonical genes—Fos and Bcl2—which are well recognised for their contribution to cell proliferation and survival. Analysis of the early mouse embryo showed that these two genes are transcribed transiently at the time of embryonic genome activation (Jin and O’Neill 2007, 2010, 2011). Detailed analysis showed that Paf induced the transcription of both these genes in the 2-cell embryo and was dependent upon the Paf-receptor, PI3K and calcium signalling (Jin and O’Neill 2011). Paf generates calcium transients with a periodicity of around 90–120 min and transcription of Bcl2 and Fos seems to occurs with a similar periodicity (Jin and O’Neill 2011). The transcripts lasted for around 40 min. By contrast both BCL2 and FOS protein have much longer half-lives (8–20 and 2–4 h, for respectively), and both are maintained at high levels throughout the pre-implantation phase (Jin and O’Neill 2011). Pharmacological inhibition of BCL2 (HA14–1) induced an increased rate of apoptosis in the resulting embryos, which was partially offset by the addition of exogenous Paf to media (Jin and O’Neill 2011). Thus, transient and periodic activation of trophic signalling pathways induces pulses of transcription of key proto-oncogenes which then results in the prolonged and persistent expression of proteins necessary for normal embryo survival (Jin and O’Neill 2011). These results provide a mechanism whereby activation of autocrine trophic signalling at fertilisation induces activation of embryonic transcription which generates a pro-survival transcriptome capable of persisting across critical transitions during early embryo development.
A role for BCL2 is the sequestration and inhibition of the pro-apoptotic protein BAX. BAX is commonly found to be expressed in embryos subjected to stress and is increased in embryos exposed to trophic factor deprivation (Chandrakanthan et al. 2006; Chi et al. 2000). The expression of BAX is canonically under the regulation of the tumour suppressor protein 53 (TRP53) transcription factor, implicating this tumour suppressor protein in embryo survival. BAX (Chin et al. 2009) and TRP53 (Jin et al. 2009) expression can be repressed in vitro by trophic ligands (discussed below).
While FOS and BCL2 are canonical response gene products to CREB , it is important to be mindful that activated CREB recognises and binds to CREB response elements (CRE) in many genes The CRE sequence is one of the most common of all response elements within the promoters of genes (Walton and Dragunow 2000; Zhang et al. 2005). Systematic genome-wide analyses suggests that CREB targets around 4084 human loci, with the total number of CREs in the genome being 750,837 (Zhang et al. 2005). Importantly, 332 of a total 866 human transcription factor genes are putative CREB targets (Zhang et al. 2005). The richness of CREB’s potential targets means that CREB activation has the capacity to not only alter the transcriptome directly, but to also create secondary effects by its capacity to recruit the expression of new transcription factors . By such mechanisms, autocrine signalling in the embryo may have the capacity to induce a significant transformative elaboration of the embryonic transcriptome.
5.8 Other PI3K-Mediated Survival Signalling Events
Pharmacological inhibition of AKT did not block the PI3K-dependent activation of calcium transients in the early embryo induced by Paf (Li et al. 2007b). This result indicates that the PDK1/AKT and calcium responses to PI3K induced signalling in the embryo create independent downstream signalling pathways. PDK1/AKT seem to have roles in activation of the transcriptional machinery required for embryonic genome activation and perhaps the MPF required for the second mitotic division, while the calcium signalling pathway has roles in the activation of essential transcription factors such as CREB . This indicates a role for autocrine embryotrophins in providing a functional link between fertilisation and the activation of the embryonic genome, and the formation of a pro-survival transcriptome. However, the activation of AKT is also known to have many targets independent of any putative actions associated with transcriptional activation (Fig. 5.2). This includes: (1) the activation of MDM2 , an important ubiquitin protein ligase (the net action of MDM2 activation is the degradation of the pro-death mediator, TRP53 (Mayo and Donner 2001), which results in TRP53 being maintained at very low levels within normal cells); (2) the inhibition of GSK3 (Fang et al. 2000), its kinase activity potentially targets glucose metabolism (Grimes and Jope 2001), the Wnt signalling pathway (Wu and Pan 2010) and the NFAT transcription factor pathway (Beals et al. 1997); and (3) the inhibition of the pro-apoptotic mediator BAD (Kulik and Weber 1998). Activation of a range of these pathways is involved in the normal survival of the preimplantation embryo (see review—(O’Neill 2008). The most detailed analysis has been performed on the roles of survival signalling in the repression of TRP53 action by AKT-mediated MDM2 activation in the early embryo.
5.9 Survival Signalling Induces Latency of TRP53
The activation of AKT down-stream of embryotrophic survival signals causes phosphorylation of serine-166 in MDM2. This activation of MDM2 is required to keep TRP53 in a latent sate within the embryo (Jin et al. 2009). Blocking the action of MDM2 pharmacologically (Nutlin-3) (Jin et al. 2009) or by the genetic deletion of Mdm2 (Jones et al. 1995; Montes de Oca Luna et al. 1995) caused TRP53-dependent death of the early embryo. The genetic deletion of the Paf-receptor or the pharmacological inhibition of PI3K or AKT in vitro also caused TRP53 accumulation and the loss of embryo viability (Jin et al. 2009; Lu et al. 2004). Importantly, TRP53 also acts to induce the transcription of MDM2 . Consequentially, there is a feedback loop between these two regulators resulting in TRP53 being tightly regulated (Moll and Petrenko 2003). A failure of trophic signalling therefore causes reduced activation of MDM2 leading to the failure of TRP53 degradation and its accumulation within cells (Jin et al. 2009). Since TRP53 is a transcription factor it can drive its own pattern of transcription including the expression of BAX and the potential resultant loss of cells due to apoptosis . It is likely that reducing survival signalling shifts the embryonic transcriptome from one generated by CREB to that generated by TRP53.
Embryos produced by IVF or those cultured from the early zygote stage are likely to have an increased presence of TRP53 within their cells, which becomes most apparent by the morulae to blastocyst stage (Chandrakanthan et al. 2006, 2007; Jin et al. 2009; Li et al. 2007a). TRP53 accumulates within the nuclei of blastocysts, indicative of its functional activation. This is confirmed by the TRP53-dependent accumulation of BAX in the early embryo after culture or IVF (Chandrakanthan et al. 2006). In the mouse, there are significant differences between strains in their susceptibility to culture and there is a direct association between this susceptibility and the accumulation of TRP53 in embryos (Ganeshan et al. 2010; Li et al. 2007a). The critical role for TRP53 in the loss of viability of embryos suffering trophic deprivation is amply illustrated by the marked improvement in the viability of embryos that are susceptible to the adverse affects of culture when TRP53 was genetically deleted or pharmacologically inhibited (Jin et al. 2009). This improvement was not so evident in a mouse strain (B6CBF1) that was resistant to loss of viability in vitro (Jin et al. 2009; Li et al. 2012). In this resistant strain the inhibition of PI3K, AKT or MDM2 all induce increased TRP53 accumulation and a resulting loss of embryo survival, indicating that in this strain PI3K-mediated trophic signalling is still required for maintenance of TRP53 latency, but that the culture conditions used did not limit survival signalling sufficiently to break this latency (Jin et al. 2009). These results show the critical role of trophic signalling pathways via PI3K in maintaining TRP53 in a latent state for the normal survival of the pre-implantation embryo.
This survival signalling also has longer term consequences. IVF and culture of embryos in well controlled conditions normally allows for a high rate of formation of morphological blastocysts. Yet a relatively small (but variable) proportion of these blastocysts have the capacity to form viable fetuses and offspring (Li et al. 2007a). This infers that some cellular or molecular ‘memory’ exists within the embryo that subsequently compromises their capacity for exercising their full developmental programme. The accumulation of TRP53 in the early embryo may account for a significant amount of this ‘memory’. When embryos were cultured from the zygote stage through to the blastocyst stage and the morphological blastocysts transferred to the uterus of recipient females, differences in the rate of development of normal fetuses was markedly influenced by the Trp53 gene dosage of the embryo. Those cultured blastocysts that lacked Trp53 had a 3–4 fold higher rate of successful fetal development after embryo transfer than their wildtype sibling embryos (Li et al. 2007a).
Detailed analysis of the impact of Trp53 on the long-term viability of cultured embryos showed that there was a similar rate of implantation of wildtype embryos as Trp53 -/- blastocysts, yet a larger proportion of the implanted wildtype embryos failed to develop to normal fetuses than was the case for Trp53 −/− implanted embryos (Li et al. 2007a). One interpretation of the higher incidence of anembryonic pregnancy of transferred cultured embryos in the presence of Trp53 is that there is a difference in the response to the accumulation of TRP53 by the cells destined to form the trophoblastic lineage (and eventually the placenta) and those destined to form the epiblast (and the embryo proper).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


