Sterilization and Pasteurization in Healthcare Facilities
Lynne M. Sehulster
Walter W. Bond
The findings and conclusions in this chapter are those of the authors and do not necessarily represent the official position of the Centers for Disease Control and Prevention. Any use of mention of trade names in this chapter is for identification purposes only and does not represent any endorsement by either the CDC or the U.S. Public Health Service.
Since ancient times, man has understood the importance of heat and water as purifying agents. One of the first references to purifying rituals using heat and water is found in the Old Testament, describing the cleaning procedures for warriors’ blood-contaminated weapons and clothing after the battle:
“Purify every garment, everything made of leather, everything woven of goat’s hair, and everything made of wood. Only the gold, the silver, the bronze, the iron, the lead, everything that can endure the fire, you shall put through the fire, and it shall be clean; and it shall be purified with the water of purification.1 But all that cannot endure fire you shall put through water.”
Numbers 31:20, 22-23 (New King James Version)
Although mankind held a vague concept that an unseen presence was associated with the spread of disease, scientific awareness and acceptance of the microbial world emerged in Europe beginning in 1677 with Anton van Leeuwenhoek’s invention of the microscope. Over the next 300 years, the great names in microbiology and medicine (e.g., Lazzaro Spallanzani, Louis Pasteur, and Robert Koch) advanced our understanding of the microbial world, all of which instituted efforts in the civilized world to develop the means to control infectious diseases through environmental sanitation, personal hygiene, and processes to preserve and protect food and water. The canning industry was among the first to employ pressurized steam sterilization to inactivate spoilage microorganisms, thereby rendering containerized food products safe to eat even after long periods of storage. In fact, Geobacillus stearothermophilus (formerly Bacillus stearothermophilus), the source of the highly resistant bacterial endospores used today as the reference standard in the design and monitoring of steam autoclave cycles, is a common food spoilage microorganism.
It has been little more than 110 years since the development of formal procedures for the sterilization of instruments and medical devices, liquids, and other materials used in hospitals. At that time, there was a growing appreciation for the role of microorganisms in the transmission of infectious diseases and for the necessity of sterile materials in surgery and other hospital activities. Although infection prevention as we know it today was in its infancy, scientists both in hospitals and in the medical device industry began to sterilize instruments and materials used in patient treatments (1).
Historically, physical methods (e.g., steam under pressure in steam autoclaves, dry heat in sterilizing ovens) have played the predominant role for sterilizing devices, equipment, and supplies in hospitals and industry. Since the 1950s, however, the number of heat-sensitive devices that must be sterilized using low-temperature processes has increased. Those low-temperature sterilization processes available to hospitals may employ gases, gas plasmas, or liquid chemicals, whereas the medical device industry typically uses gas (ethylene oxide, ETO) or irradiation for terminal sterilization. Exposure of heat-sensitive materials to short-cycle substerilizing temperatures (e.g., pasteurization [60-75°C for 5-15 minutes]) is common in the food industry but of limited use in the medical arena in the United States.
The material in this chapter centers on a review of the general principles of sterilization and pasteurization as currently performed primarily in US healthcare facilities. Discussion of the sterilization methods used in industry (e.g., irradiation), as well as an in-depth review of the mathematical/engineering basis of sterilization, is beyond the scope of this chapter. The reader is referred to two excellent texts for more detailed information on these topics (2,3). This chapter also does not discuss the use of liquid chemicals for disinfection as this topic is thoroughly covered in Chapter 80. Furthermore, the day-to-day operations and maintenance of sterilizers and all the attendant processes, occupational safety, construction, regulations, and quality control aspects, are addressed in Chapter 70.
DEFINITIONS
Over the years, the terms “sterilization,” “sterile,” “decontamination,” and “disinfection” have often been used
interchangeably, thereby leading to considerable confusion among healthcare personnel. For “sterile” and “sterilization,” we view these, respectively, in the context of the state of sterility, as the process of sterilization and the application of sterilization. Any device or solution is considered to be sterile when it is completely free of all microorganisms, either living or inert (i.e., viruses, bacterial endospores, or fungal spores) (1). This state of sterility is the objective of the sterilization procedure, and viewed in this context, the definition is categorical and absolute (i.e., an item is either sterile or it is not). An interesting dilemma arises with fluids “sterilized” via filtration, using bacteriologic filters (nominal pore size of 0.2 µm) for this purpose. Such filtered liquids are often labeled as “sterile,” but the pore size of the filters used is large enough to permit the passage of viruses or certain pleomorphic or cell wall-deficient gramnegative bacterial species (e.g., species of Pseudomonas, Proteus, and Stenotrophomonas), Mycoplasma spp., or Mycobacterium spp. (including Mycobacterium tuberculosis and waterborne nontuberculous mycobacteria) to the filtrate. Therefore, in the most practical sense of the term, such liquids do not meet the strict definition of “sterile” (4). This is especially true if the fluid being filtered has a potentially high microbial content (e.g., untreated, contaminated water or, not infrequently, water from the piping and tubing systems of treated public/institutional water supplies). An exception is noted for the end-stage filtration of “finished” liquids in the production of pharmaceutical and medical products that have been subjected to one or more rigorous prefiltration purification steps.
interchangeably, thereby leading to considerable confusion among healthcare personnel. For “sterile” and “sterilization,” we view these, respectively, in the context of the state of sterility, as the process of sterilization and the application of sterilization. Any device or solution is considered to be sterile when it is completely free of all microorganisms, either living or inert (i.e., viruses, bacterial endospores, or fungal spores) (1). This state of sterility is the objective of the sterilization procedure, and viewed in this context, the definition is categorical and absolute (i.e., an item is either sterile or it is not). An interesting dilemma arises with fluids “sterilized” via filtration, using bacteriologic filters (nominal pore size of 0.2 µm) for this purpose. Such filtered liquids are often labeled as “sterile,” but the pore size of the filters used is large enough to permit the passage of viruses or certain pleomorphic or cell wall-deficient gramnegative bacterial species (e.g., species of Pseudomonas, Proteus, and Stenotrophomonas), Mycoplasma spp., or Mycobacterium spp. (including Mycobacterium tuberculosis and waterborne nontuberculous mycobacteria) to the filtrate. Therefore, in the most practical sense of the term, such liquids do not meet the strict definition of “sterile” (4). This is especially true if the fluid being filtered has a potentially high microbial content (e.g., untreated, contaminated water or, not infrequently, water from the piping and tubing systems of treated public/institutional water supplies). An exception is noted for the end-stage filtration of “finished” liquids in the production of pharmaceutical and medical products that have been subjected to one or more rigorous prefiltration purification steps.
Sterilization is the use of a physical, chemical (liquid, gas, or plasma), or irradiation process to destroy all microorganisms including large numbers of highly resistant bacterial endospores. A sterilization procedure, on the other hand, cannot be categorically defined. Rather, the procedure is defined as a process, after which the probability of a microorganism surviving on an item is <1 in 1 million (10 and 11). This is referred to as the sterility assurance level (SAL), and the attendant concepts are used by the medical device industry to design and monitor sterilization cycles for wide varieties and large quantities of medical devices.
The application of sterilization takes into account additional considerations (e.g., the Spaulding terminology used in the classification of materials subjected to various germicidal processes). Dr. E. H. Spaulding in 1972 (5) devised a classification scheme for reusable medical devices and instruments to determine the appropriate terminal reprocessing step for each of these items. Such medical devices and instruments were categorized as “critical,” “semi-critical,” or “noncritical” based on the manner in which they come into contact with patients and the corresponding risk of infection transmission during their use. Critical instruments or devices, the first category, are so called because of the substantial risk of infection if the item is contaminated with microorganisms at the time of use (i.e., with these instruments, sterility is critical). These are instruments or objects that are introduced directly into the human body, either into or in contact with the bloodstream or normally sterile areas of the body. Examples include needles, scalpels, transfer forceps, cardiac catheters, implants, and also the inner surface components of extracorporeal blood-flow devices, such as those of the heart-lung oxygenator and the blood side of artificial kidneys (hemodialyzers). These items must be meticulously cleaned, then sterilized by a validated sterilization process before use on the next patient, thereby minimizing the potential for disease transmission. Instruments or devices in the second category are classified as semi-critical in terms of the risk of infection, and examples are flexible gastrointestinal endoscopes, bronchoscopes, endotracheal and aspirator tubes, respiratory therapy equipment, and vaginal specula. Although these items come in contract with mucous membranes, they do not ordinarily come into contact with the bloodstream or a sterile body site during normal use. Sterilization of many of these items, although desirable and cost-effective if they can withstand steam autoclaving, is not absolutely essential (i.e., according to Spaulding’s classification, it is semi-critical that they be sterile at the time of use). At a minimum, semi-critical instruments or devices should be subjected to the comparatively potent heat or chemical treatment referred to as high-level disinfection— a process that is discussed in detail in Chapter 80. Instruments, devices, or surfaces that come into direct or indirect contact only with intact skin during routine use (e.g., backboards, blood pressure cuffs, and stethoscopes) have only minimal risk of disease transmission. The sterility of these surfaces and devices is noncritical. For some of these items and surfaces, washing with soap and water may be the only between-use process necessary. Medical equipment used routinely in patient care is typically cleaned and disinfected routinely, but care must be used to minimize damage to and malfunction of electronic medical equipment when using liquid cleaners and disinfectants. Cleaning and application of low- to intermediate-level disinfection for noncritical surfaces are defined and discussed in Chapter 80.
GENERAL FACTORS AFFECTING STERILIZATION PROCESSES
A significant consideration in assessing the effectiveness of a sterilization process is the overall resistance level of the target microbial population. Resistance can be categorized as intrinsic or extrinsic. Intrinsic resistance is the innate capacity of a particular microbial cell to resist a specific potentially lethal treatment. Extrinsic resistance, in contrast, describes the added challenge that inert organic matter and other extraneous materials or configurations (e.g., mated surfaces such as forceps’ hinges) pose to a sterilizing process. Organic matter, soil, patient material, or charred matter can diminish the effectiveness of the sterilizing treatment. It follows that high numbers of the most highly resistant microorganisms and large amounts of organic matter present on a device or surface will contribute significantly to the overall resistance of the population to inactivation and therefore, both of these contaminants should be minimized prior to sterilization or any other terminal reprocessing. Meticulous cleaning of devices, instruments, and surfaces is therefore an enormously important first step that will ensure the overall success of the process.
Intrinsic Resistance
Microorganisms vary widely in their responses to physical and chemical stresses. Among the various categories of
microorganisms, the species that form bacterial endospores possess the highest level of intrinsic resistance to sterilization processes, and because of this, they are consistently used as standard challenge microorganisms when initially designing and engineering a sterilization cycle and also when monitoring in-use performance of sterilizer equipment in the healthcare setting. Intrinsic resistance stems from innate and specific biophysical and biochemical properties of the particular strain of the spore-forming bacterium. These properties can be affected by external factors such as pH and the ionic concentration of growth and sporulation media, all of which can have pronounced effects on spore resistance to heat. Endospores are more readily inactivated at low pH because pH can affect ionic adsorption to the spore surface, which in turn can affect heat stability (6). In contrast, yeasts in low pH media can exhibit increased resistance to heat (2). With respect to thermal resistance of bacterial endospores, Powell, in 1953, identified dipicolinic acid (DPA) (pyridine-2, 6-dicarboxylic acid) as closely associated with thermoresistance (7). This chemical is found in correspondingly high levels in all endospores with significant resistance to heat, but is absent in the vegetative bacterial cells (8). When sterilization researchers developed the inactivation kinetic rates (survival curves) for various bacterial endospores, they observed that endospores first emerge from a dormant state, and the cellular energy required to do this is called “activation energy.” This is the initial step in the inactivation process and also occurs prior to cellular growth. Grecz et al., in 1972, determined that this step involved the dissolution of a calcium salt of DPA. The rate of DPA loss is proportional to levels of intrinsic thermal resistance of the endospores (9). Endospores of different genera and species often demonstrate widely differing levels of intrinsic resistance to different sterilizing agents. A classic example of this observation is seen with heat applications. For moist heat sterilizing processes, endospores of G. stearothermophilus provide the greatest challenge for this process, whereas their resistance to dry heat is very low. Bacillus atrophaeus (formerly B. subtilis var. niger) endospores have greater resistance to dry heat than to moist heat (3). Another example illustrates this point. A curious, extremely slow-growing bacterium, Bacillus xerothermodurans, was isolated from soil collected near the National Aeronautics and Space Administration (NASA) Vehicle Assembly Building (VAB) at Cape Kennedy, FL, in the 1970s (10,11). Endospores of this microorganism exhibited an extreme level of intrinsic resistance to dry heat (i.e., at 125°C, the length of time needed to reduce the number of surviving spores by 90% was 139 hours). In contrast, endospores of B. atrophaeus, the standard challenge microorganism used to design and monitor dry heat or ETO sterilization cycles, showed a 90% reduction of surviving spores when exposed for 15 minutes to dry heat at 125°C. In other words, B. xerothermodurans is >500-fold more resistant to inactivation by dry heat than is B. atrophaeus. Interestingly, endospores of B. xerothermodurans are extremely sensitive to inactivation with moist heat (i.e., a 90% reduction of surviving spores occurs when exposed to moist heat at 80°C for 61 minutes). Although serving as an illustration of the spectrum of spore resistance, these contrasting extremes offer little practicality for this novel microorganism being used as a biological indicator. It may, however, eventually serve as a useful research tool in the ongoing study of microbial resistance to heat.
microorganisms, the species that form bacterial endospores possess the highest level of intrinsic resistance to sterilization processes, and because of this, they are consistently used as standard challenge microorganisms when initially designing and engineering a sterilization cycle and also when monitoring in-use performance of sterilizer equipment in the healthcare setting. Intrinsic resistance stems from innate and specific biophysical and biochemical properties of the particular strain of the spore-forming bacterium. These properties can be affected by external factors such as pH and the ionic concentration of growth and sporulation media, all of which can have pronounced effects on spore resistance to heat. Endospores are more readily inactivated at low pH because pH can affect ionic adsorption to the spore surface, which in turn can affect heat stability (6). In contrast, yeasts in low pH media can exhibit increased resistance to heat (2). With respect to thermal resistance of bacterial endospores, Powell, in 1953, identified dipicolinic acid (DPA) (pyridine-2, 6-dicarboxylic acid) as closely associated with thermoresistance (7). This chemical is found in correspondingly high levels in all endospores with significant resistance to heat, but is absent in the vegetative bacterial cells (8). When sterilization researchers developed the inactivation kinetic rates (survival curves) for various bacterial endospores, they observed that endospores first emerge from a dormant state, and the cellular energy required to do this is called “activation energy.” This is the initial step in the inactivation process and also occurs prior to cellular growth. Grecz et al., in 1972, determined that this step involved the dissolution of a calcium salt of DPA. The rate of DPA loss is proportional to levels of intrinsic thermal resistance of the endospores (9). Endospores of different genera and species often demonstrate widely differing levels of intrinsic resistance to different sterilizing agents. A classic example of this observation is seen with heat applications. For moist heat sterilizing processes, endospores of G. stearothermophilus provide the greatest challenge for this process, whereas their resistance to dry heat is very low. Bacillus atrophaeus (formerly B. subtilis var. niger) endospores have greater resistance to dry heat than to moist heat (3). Another example illustrates this point. A curious, extremely slow-growing bacterium, Bacillus xerothermodurans, was isolated from soil collected near the National Aeronautics and Space Administration (NASA) Vehicle Assembly Building (VAB) at Cape Kennedy, FL, in the 1970s (10,11). Endospores of this microorganism exhibited an extreme level of intrinsic resistance to dry heat (i.e., at 125°C, the length of time needed to reduce the number of surviving spores by 90% was 139 hours). In contrast, endospores of B. atrophaeus, the standard challenge microorganism used to design and monitor dry heat or ETO sterilization cycles, showed a 90% reduction of surviving spores when exposed for 15 minutes to dry heat at 125°C. In other words, B. xerothermodurans is >500-fold more resistant to inactivation by dry heat than is B. atrophaeus. Interestingly, endospores of B. xerothermodurans are extremely sensitive to inactivation with moist heat (i.e., a 90% reduction of surviving spores occurs when exposed to moist heat at 80°C for 61 minutes). Although serving as an illustration of the spectrum of spore resistance, these contrasting extremes offer little practicality for this novel microorganism being used as a biological indicator. It may, however, eventually serve as a useful research tool in the ongoing study of microbial resistance to heat.
Extrinsic Resistance
The presence of soil, organic matter, and/or bioburden on the surface of a medical device or instrument will interfere with the ability of the sterilant to make contact with those surfaces, thereby interfering with and diminishing the effectiveness of the sterilization process. The protective effect of organic matter has been recognized for several decades. With respect to moist heat (e.g., steam under pressure) sterilizing processes, the presence of residual organic matter can extend the time to inactivate bacterial endospores by six to eight times compared with the time required to inactivate spores in a less organically contaminated milieu (12). Further, organic bioburden not only provides physical protection but may also contain massive numbers of microorganisms with widely varying levels of resistance, thereby increasing the potential for highly resistant microorganisms to be present. Bioburden and inorganic matter can pose challenges for other sterilizing processes as well. Dry heat is an oxidative process, and anything that prevents oxygen from reaching its targets will confer resistance (3,13,14). For example, the inclusion of bacterial endospores inside inorganic, water-soluble crystals can extend the dry-heat inactivation times by 15-fold to 20-fold (15).
Cleaning
Cleaning is a process using water and any of a variety of salts, surfactants, ultrasonic energy, or enzymes to break covalent bonds between surfaces and visible soils, organic matter, and microorganisms, thereby enabling the dispersion and removal of these foreign materials from the item. Cleaning is, therefore, absolutely essential in efforts to enhance the efficacy of any terminal reprocessing step and is equally important for both sterilization and disinfection processes (4). A number of instrument design factors can pose challenges to effective cleaning. These include the presence of lumens, closely mated surfaces, acute angles, springs and valves, inaccessible areas, and roughened surfaces (16). Cleaning can be done manually, removing soil via water, detergent, and physical scrubbing, or by using an automated process such as enzymatic detergents and ultrasonic cleaners. Ultrasonic cleaning involves waves of acoustic energy in liquid, using cavitation and implosion to loosen soils from surfaces (17). An efficient cleaning process is capable of reducing microbial bioburden on instrument surfaces by several orders of magnitude. A number of researchers have demonstrated that microbial bioburden on typical medical instruments is generally low (<103 colony-forming units [CFU]/device) before cleaning, but ≥80% will have <102 CFU/device after cleaning (18, 19 and 20). In one series of experiments, Rutala et al. (20) observed that approximately 30% of cleaned medical instruments had no detectable contamination by the sampling and culture methods being used. Some of the microorganisms isolated from cleaned instruments have included coagulase-negative Staphylococcus aureus, diphtheroids, Bacillus cereus, Bacillus spp., and Stenotrophomonas maltophila. This observation supports the notion that while cleaning is essential to the overall efficacy of any terminal reprocessing, cleaning by itself is not by any means sufficient
to render a critical medical device or instrument safe and ready for use on the next patient. Such devices and instruments must always be subjected to a validated sterilization process prior to use.
to render a critical medical device or instrument safe and ready for use on the next patient. Such devices and instruments must always be subjected to a validated sterilization process prior to use.
This question has been asked on many occasions—is it safe to use a reusable medical device with residual foreign material after the device has undergone a sterilization process? The risk of infection in this instance is difficult to estimate, but the current consensus is that this questionable work practice (the state of “sterile but dirty”) does not meet a proper surgical standard. The proper course of action in this instance is to return the instrument or lot of instruments to the central sterile supply unit (CSS) to be recleaned and resterilized. Even if the risk of infection is negligible, the possibility exists that such residual foreign matter may cause localized irritation and subsequent inflammation in the surgical site or may trigger an immune response, all of which may contribute to a possible surgical site adverse event. The aesthetics of such a lack of physical cleanliness is self-evident.
Sterility Assurance Levels and Engineering a Sterilization Cycle
Historically, the development of a sterilization cycle involved the determination of a SAL by using a specific biological challenge microorganism (e.g., representative bacterial endospores of significantly high resistance), constructing an inactivation curve, and obtaining a D-value. Death in this instance is a first order exponential or logarithmic function. In general, a D-value is the time required at a specific temperature to inactivate a given microbial population by 90% (i.e., a one log10 reduction, examples of which were given earlier). As mentioned previously, activation energy is required to bring spores out of dormancy as the very first step of inactivation, and when plotted in an inactivation curve, this may distort the initial shape of the curve depending on the innate characteristics of the endospore population being tested (3). D-values, therefore, are valid only when treatment temperatures are high enough to discount the effect of activation and the population of challenge microorganisms is of uniform resistance (3). This approach is illustrated in Figure 81-1. The inactivation rate is linear, and the D-value can be obtained for a given set of sterilization parameters. In practice, a survivor curve spanning six logs of inactivation can be determined by using quantitative spore assays. A sixlog 10 reduction in this discussion is considered a half cycle (1). An additional six log10 inactivation can be deduced by extrapolation of the linear survivor curve. In other words, the time at temperature required for the half cycle is doubled, resulting in a 12-log10 level of lethality for a full cycle. This procedure is used to construct a sterilization cycle with an SAL of 10-6, meaning that the probability of a single survivor in the initial challenge of highly resistant endospores would be 1 in 1 million (21). In the development of sterilization cycles, this approach is extraordinarily conservative. Accordingly, manufacturers of sterilization systems, including low-temperature systems, employ this high degree of conservatism in designing their cycles (Box 81-1).
In reality and as mentioned previously, most reusable medical devices have a bioburden <102 CFU/device after cleaning, and the residual microorganisms consist primarily of vegetative bacteria that are killed much more rapidly than are bacterial endospores. In the design and validation of the sterilization cycle, a challenge of one million bacterial endospores highly resistant to the process is used, and the survival curves reflect the highly conservative nature of this approach. This design conservativism, in conjunction with consistently efficient instrument and device cleaning, results in a significant level of overkill, thereby ensuring a huge margin of sterilization success and a high degree of patient safety (1,22).
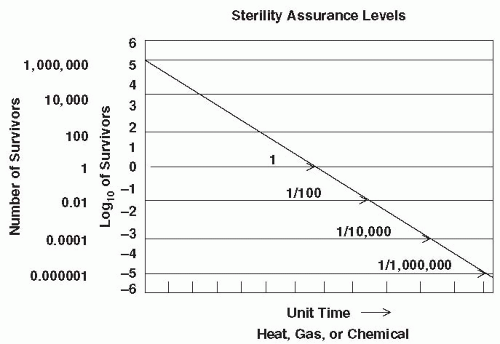 FIGURE 81-1 Numbers of surviving microorganisms compared to a starting population of 1,000,000 microorganisms are as follows: 6 logs10 = 1,000,000; 5 logs10 = 100,000; 4 logs10 = 10,000; 3 logs10 = 1,000; 2 logs10 = 100; 1 log10 = 10; 0 logs10 = 1. Three examples of the probability of a surviving microorganism as inactivation time continues are noted in the chart. (Adapted from Figure 43.2 in Favero MS, Bond WW. Chemical disinfection of medical and surgical materials. In: Block SS, ed. Disinfection, sterilization, and preservation. 5th ed. Philadelphia, PA: Lippincott, Williams and Wilkins, 2001:881-917.) |
When engineering a sterilization cycle, regardless of the type of sterilant used (e.g., steam under pressure, gas plasma, and dry heat), researchers will take the following parameters into account: time, temperature, relative humidity (RH), pH of any suspending medium used with challenge microorganisms, and a standardized load configuration. Time and temperature, as mentioned previously, are the basic parameters evaluated when constructing inactivation curves and determining D-values. RH is defined as the ratio of the actual water vapor pressure in a system
to the saturated water vapor pressure of the system at the same temperature. Water activity is the relative water availability in a cell or spore and depends on the RH (23). There appears to be an inverse relationship between cell resistance and water activity (23). The more water available to cells or spores, the faster the inactivation process whether it be moist heat or dry heat (24).
to the saturated water vapor pressure of the system at the same temperature. Water activity is the relative water availability in a cell or spore and depends on the RH (23). There appears to be an inverse relationship between cell resistance and water activity (23). The more water available to cells or spores, the faster the inactivation process whether it be moist heat or dry heat (24).
BOX 81-1
Sterilization Cycle Design Considerations
An assumption that the bioburden on a device is 106, consisting of bacterial endospores most resistant to the process
In challenge tests, the placement of endospores is in the most occluded location in the device
Use of spores contained in an organic and inorganic burden
Application of simulated use conditions
Documentation that a complete cycle produces a six log10 of measurable kill with a 10-6 probability that one spore survives
Standardized sterilizer loading configurations are important for a number of reasons. Regardless of the process used, the sterilant must be able to make contact with all surfaces in the load in order to effect microbial inactivation. Load configurations take into account the packaging of devices, placement in secondary containment, etc., in efforts to identify spatial challenges to sterilant penetration. Such challenges can include “cold spots” in the load where temperature is less than that specified for the cycle and air pockets due to insufficient air evacuation processes. Once the manufacturer of the sterilizer has validated the sterilization cycle parameters and confirmed effectiveness with standardized loads, in-use instructions can then ensure that hospitals will be able to use the equipment effectively and safely.
STERILIZATION PROCESSES
In the United States, the Food and Drug Administration (FDA) recognizes three categories of sterilization methods used to sterilize medical devices in manufacturing settings. As described in draft guidance for industry, these categories are (a) traditional sterilization methods, (b) nontraditional sterilization methods, and (c) novel nontraditional sterilization methods (25). Traditional sterilization methods are those that have a long history of safe and effective use as demonstrated by ample literature, 510(k) clearances or approvals of premarket approval applications, and satisfactory quality systems (QS) inspections, and for which there are voluntary consensus standards for validation that are recognized by the FDA (25). Sterilization methods in the traditional category include dry heat, ETO in a fixed chamber, moist heat, and irradiation (e.g., gamma ray, electron beam). Nontraditional sterilization methods are those that do not have a long history of safe and effective use and for which there are no FDA-recognized standards but for which published information on the validation of these methods exists and for which the FDA has previously evaluated data as part of a QS evaluation and determined the methods to be adequate (25). Hydrogen peroxide gas plasma and ozone are the two methods currently listed under the nontraditional category. Of the six sterilization methods/sterilants listed in these two categories, all with the exception of irradiation are commonly available for use in US healthcare facilities. ETO, hydrogen peroxide gas plasma, and ozone are available in CSS settings as low-temperature processes for heat-sensitive instruments. Novel, nontraditional sterilizing methods are newly developed methods for which there are no FDA-recognized standards, no FDA inspection history, and no FDA comprehensive evaluations of sterilization validation data (25). There is also little or no published information on process validation for these methods. Furthermore, a novel, nontraditional method is one that has not been evaluated by the FDA as part of a QS evaluation and that employs sterilization methods that the FDA has not reviewed and determined to be adequate to provide a reasonable assurance of safe and effective use. Methods included in this category are chlorine dioxide, ETO in a bag, high-intensity light or pulsed light, microwave irradiation, sound wave energy, ultraviolet light, and vaporized chemical sterilant systems (e.g., hydrogen peroxide or peracetic acid [PAA]) (25). These novel, nontraditional methods/sterilants will not be addressed in this chapter, given their current status as emerging, developing technologies.
It is interesting to note that sterilization using liquid chemical sterilants is not included in this FDA discussion of sterilization methods. A liquid chemical sterilization process is by design problematic, time-consuming, and has no procedure or method to ensure or maintain the sterility of the device once it has been removed from the chemical. One possible explanation for the omission is that the draft guidance containing the definitions for the three sterilization method categories is intended for the device industry, and device manufacturers would be asked to identify the sterilization method used to render devices sterile before marketing. In view of the fact that liquid chemical sterilization methods involving device immersion do require long contact times and there is a risk of contamination of the devices upon removal from the chemical, it is unlikely that device manufacturers would employ a liquid chemical sterilization method. Liquid chemical sterilization, however, is a method available for use in the healthcare setting (i.e., the device customer) for the reprocessing of reusable, heat-sensitive devices.
STERILIZATION BY HEAT
Heat-based sterilization processes in healthcare facilities are based on either moist heat (steam under pressure) or, to a lesser degree, dry heat.
Moist Heat (Saturated Steam under Pressure)
Moist heat technology for terminal reprocessing of heatstable reusable medical instruments and devices is the most common, efficient, and economical sterilization process in use. Furthermore, as a properly conducted sterilizing process, it leaves no chemical residues or byproducts on the surfaces of the medical instruments. Its lethal property is the result of mass transfer of heat to a surface via convection, with steam condensing as it loses heat. Thus, steam kills quickly and efficiently and does this by coagulation and denaturation of enzymes and structural proteins (17). The basic steam sterilizer (autoclave) is a steel-walled chamber surrounded on the sides by a jacket of trapped steam. The autoclave chamber may be sealed at the back of the chamber, or it may be designed as a “pass-through” chamber with doors located at either end. Pressure gauges, safety relief valves, temperature indicators, steam entry portals, and drain vents are some of the main engineered components of this equipment.
One of the major concerns of autoclave design and engineering is the removal of air from the load and the chamber. Although there are several different engineering approaches to air removal, the two main mechanisms used to accomplish air removal are gravity displacement and air evacuation (3). There are three types of general-purpose
steam sterilizers that use these methods of air removal: (a) gravity displacement sterilizers, (b) dynamic airremoval sterilizers, and (c) steam-flush pressure pulse sterilizers (26). Equipment employing vacuum evacuation of air was previously referred to as “prevacuum” or “porous load” sterilizers, but the Association for the Advancement of Medical Instrumentation (AAMI) now identifies these as dynamic air-removal sterilizers (27).
steam sterilizers that use these methods of air removal: (a) gravity displacement sterilizers, (b) dynamic airremoval sterilizers, and (c) steam-flush pressure pulse sterilizers (26). Equipment employing vacuum evacuation of air was previously referred to as “prevacuum” or “porous load” sterilizers, but the Association for the Advancement of Medical Instrumentation (AAMI) now identifies these as dynamic air-removal sterilizers (27).
Gravity displacement autoclaves are typically used to sterilize nonporous instruments and devices, laboratory media, liquids, and pharmaceutical products; for in-house sterilization of glassware, oils, or powders, see “Dry Heat” below (17). This type of autoclave can range in size from small, tabletop units commonly used in small clinical practice to larger-sized units suitable for CSS operations (26). When engineered on a very large scale, this type of sterilizer is also one of the most commonly used technologies for the decontamination of regulated medical waste. Gravity displacement autoclaves are generally not recommended for terminal reprocessing of dense, porous items because the gravity displacement method of air removal in this situation is very inefficient. Entrapped air in a porous load leads to reduced steam penetration, and subsequently, heating efficiency (17,28,29).
In the “conditioning” phase of the sterilization cycle for a gravity displacement autoclave, steam enters the chamber from a portal at the top of the chamber. A water separator unit located at the steam entry portal helps to entrap water mist, removing it from the steam and thereby improving steam quality to >98% saturated steam (i.e., <2% water) and also minimizing the wetness of the load during the cycle (3). A steam separator modulates the velocity of the steam entering the chamber so as to minimize steam mixing with the air. Steam is lighter than air, and as steam fills the chamber from the top, air is forced by gravity to exit the chamber at a drain portal located on the floor of the chamber.
A variety of time, temperature, and steam pressure parameters have been validated for these sterilizers, and the reader is referred to the American National Standards Institute (ANSI)/AAMI document ANSI/AAMI ST79:2010 for suggested cycle parameters (27). Users, however, should consult the manufacturer’s written instructions for proper and effective operation of specific models of this equipment.
Dynamic air-removal autoclaves make use of a highspeed vacuum pump to remove air at the start of a cycle. Air evacuation can be accomplished in one step or by using several “pulses” of pulling a vacuum (i.e., pressure pulsing). High-vacuum air removal is a one-pulse evacuation. When the chamber pressure reaches a partial pressure <5 mm Hg, the steam is injected into the chamber, and the heating of the load is begun (3). This particular engineering design for a sterilizer has been associated with several problems, including dehydration of certain materials, generation of superheated steam in porous item packs, charring of fabric; accordingly, uses of high-vacuum air-removal sterilizers are being phased out (3). Other versions of dynamic air-removal autoclaves are designed to evacuate the air in several applications of a vacuum, all during the conditioning phase. This pressure-pulsing method of air removal is efficient at removing air entrapped in porous loads (3). Unlike gravity displacement autoclaves, dynamic air-removal sterilizers are known to have problems with air leaks and occasional inadequate air removal (17). The Bowie-Dick test is run on dynamic air-removal sterilizers at the beginning of the day to test for effective air removal (26,27). A Bowie-Dick test sheet is inserted into a porous load (e.g., clean cotton, surgical towels) and placed toward the front of the chamber over the drain. If the conditioning phase of the sterilizer fails to evacuate all entrapped air, a spot will appear on the Bowie-Dick test sheet (27). In this event, the autoclave should be taken out of service pending inspection, repairs, and a successful pass of a subsequent Bowie-Dick test (27).
A third type of general purpose steam sterilizer employs a steam-flush pressure pulse mechanism for air evacuation. Air is evacuated by drawing a vacuum to a predetermined pressure, at which point steam is introduced into the chamber, forcing air out through a vent in the chamber. This air removal process is repeated several times, and during this process, the load gradually comes to the predetermined exposure temperature (3). Unlike the dynamic air removal sterilizers, the steam-flush pressure pulse equipment is not prone to air leaks; Bowie-Dick testing is not required with this type of sterilizer (26).
As with gravity displacement sterilizers, dynamic airremoval equipment manufacturers have validated a variety of time-temperature-steam pressure combinations of parameters, and the ANSI/AAMI ST79:2010 summarizes these in thorough discussions (27). Written instructions of the sterilizer manufacturer must also be consulted when determining the appropriate cycle parameters for each type of recommended loading configuration (e.g., wrapped metal instruments or porous materials).
Sterilization using saturated steam under pressure is a demonstrably economical and effective process. However, there are three areas of concern that impact the proper operation of these pieces of equipment: (a) superheated steam, (b) load wetness, and (c) steam quality (3). Superheat is a temperature excess above the temperature of saturated steam at the same pressure (3). Superheated steam can be generated in several ways. If the temperature in the jacket is higher than the target temperature for the cycle, materials in the load can pick up this heat. When steam is introduced into the chamber, the steam picks up the heat from the load and the walls of the chamber, resulting in superheat in the surface layers of the load (3). Superheat can be deleterious to porous loads (e.g., fabrics). When heat is transferred from steam to fabric, that effect penetrates beyond the surface and into the materials deep in the load. Additionally, heat is released when moisture rehydrates dried materials (e.g., cotton fabrics, paper) (30,31). As a result, temperatures in the center of a porous load can rise and, in some instances, cause charring of the materials.
Excessive load wetness after the completion of a heat cycle can be extremely problematic. In the 1950s, researchers demonstrated that bacteria can pass through wet or damp wrapping materials, thereby contributing to postprocess contamination of the load (32,33). Most sterilizers include a drying phase as the last step of the sterilizing process to prevent this unacceptable wetness.
Steam quality is critical to the success of a steam sterilization cycle. Ideal steam for sterilization is 100% saturated
steam by weight (3). The presence of water diminishes the quality of the steam, which in turn affects the efficiency of sterilization and appropriate drying of processed materials (3). Most steam sterilizers are designed to operate most efficiently using high-quality steam (e.g., ≥97% steam). As steam makes contact with the load, heat is released to the load as the steam condenses. The water used to generate the steam is now spread out as a wet film over the entire load (34). A steam sterilizer’s dry cycle is engineered to evaporate any water that has not drained out via the chamber steam trap drains (assuming high-quality steam). Diminished quality steam adds more liquid water to the load, and if this is more than the steam sterilizer’s drying cycle can evaporate during its cycle, the load will end up with wet packs (34). Items that become extremely wet during the sterilization cycle are correspondingly difficult to dry.
steam by weight (3). The presence of water diminishes the quality of the steam, which in turn affects the efficiency of sterilization and appropriate drying of processed materials (3). Most steam sterilizers are designed to operate most efficiently using high-quality steam (e.g., ≥97% steam). As steam makes contact with the load, heat is released to the load as the steam condenses. The water used to generate the steam is now spread out as a wet film over the entire load (34). A steam sterilizer’s dry cycle is engineered to evaporate any water that has not drained out via the chamber steam trap drains (assuming high-quality steam). Diminished quality steam adds more liquid water to the load, and if this is more than the steam sterilizer’s drying cycle can evaporate during its cycle, the load will end up with wet packs (34). Items that become extremely wet during the sterilization cycle are correspondingly difficult to dry.
There are a number of causes for diminished steam quality. Boiler problems are often the first to be evaluated in this regard. Boilers must be serviced and maintained by trained personnel. The steam distribution system and piping, if not insulated, can secondarily cause condensation of steam to water en route to the sterilizers (34). Dissolved salts and other minerals in the water used to generate the steam can also affect steam quality.
Flash Sterilization At times, during surgical procedures, a medical instrument in short supply may be inadvertently dropped to the floor or somehow becomes contaminated such that it needs to be cleaned and sterilized as quickly as possible. The process of “flash sterilization” was developed to address this circumstance. Underwood and Perkins outlined the parameters of this process as the sterilization of an unwrapped item at 132°C for 3 minutes at 27 to 28 lb of pressure in a gravity displacement sterilizer (17,35). Flash sterilization is, therefore, distinguishable from the more conventional “full-cycle sterilization,” in which a clean device or instrument is wrapped and sterilized so that it may be stored and maintained in its sterile state for subsequent use (35). Historically, the Centers for Disease Control and Prevention (CDC) recommended against using flash sterilization for routine sterilization of instruments, arguing that the process should not be done as a means of convenience, to avoid having sufficient surgical instrument inventory, or to accelerate the throughput of instruments to the next surgical case (36,37). Additionally, CDC recommended at that earlier time against the use of flash sterilization for reprocessing surgical implants for the following reasons: (a) up until recently there were no rapid, reliable, validated biological indicators (BIs) available for monitoring the flash sterilization procedure; (b) there was a lack of protective wrapping to maintain the sterility of the instrument; (c) there existed a possibility of inadvertent contamination of the instrument during transport from the sterilizer to the sterile operative field in the operating room; and (d) the cycle parameters (time, temperature) were considered nearing minimal effectiveness. However, there are times when flash sterilization of an implant is unavoidable in an emergency. In this situation, the process may be optimized by paying particular attention to thorough record-keeping and documentation of the implant (lot/serial numbers, separate components, etc.), the cleaning and sterilizing parameters used, the technicians performing the reprocessing, the surgical procedure and the surgical team, patient information, day and time of the surgery, etc (17).
Carefully performed by trained personnel using modern equipment, materials, and techniques, flash sterilization can be considered an effective sterilizing process with sufficient SAL (10-6) such that potential patient-topatient transmission is interrupted with high confidence (38,39). Flash sterilization can be done on heat-stable reusable medical instruments using a steam sterilizer— either a gravity displacement unit or dynamic-air-removal equipment. The instrument(s) must first be meticulously cleaned, thoroughly rinsed, and dried. There are a variety of factors that must be taken into consideration with regard to cycle parameters before running a flash-sterilization process. These include (a) the type of steam sterilizer used; (b) the type of load (nonporous or a mix of porous and nonporous); (c) whether or not the item is wrapped or otherwise contained; (d) the temperature; (e) the exposure time; and (f) whether or not a dry-time phase is part of the cycle. The reader is referred to the ANSI/AAMI ST79:2010 (27) for summary tables of suggested cycle parameters for two configurations of steam sterilizers, but most importantly the healthcare facility wishing to use flash sterilization should consult the sterilizer manufacturer’s written instructions for information on validated cycle parameters (27). Additionally, some medical instruments and devices require longer exposure time, making it equally important to consult the manufacturers of these instruments and devices as well. Furthermore, if any sort of containment is used for the instrument or device undergoing flash sterilization, it is imperative to verify that said containment has been cleared by the FDA for this purpose, and the healthcare facility should follow the containment manufacturer’s instructions carefully. Wrappers are generally used to facilitate aseptic transport from the sterilizer to the point of immediate use after the instrument has cooled down, whereas rigid containment may be considered for aseptic holding of the instruments for short periods of time (usually <24 hours), depending on the design configuration of the container.
Recently, AAMI convened a working group of professional associations and federal government agencies (including CDC) to consider revisiting flash sterilization as practice and policy. Given the variety of cycle parameters and possible uses for shortened steam sterilization cycles, the group felt that the term “immediate-use steam sterilization” more appropriately identified this process. The working group will release a statement of its findings, but at this writing, the group continued to support these key elements: (a) manufacturer instructions for validated cycle parameters are to be followed, and containment vessels/materials should be confirmed as FDA-cleared; (b) instrument inventories should be sufficient to meet anticipated surgical demand; (c) technicians should be thoroughly trained in all aspects of performing immediate-use steam sterilization; (d) instruments must be thoroughly cleaned before sterilization; and (e) the traditional recommendations against performing immediate-use steam sterilization on implants and those instruments for which validated cycle parameters are not available are retained. An additional advisory against the use of immediate-use
steam sterilization for instruments that have been used on patients known or suspected of having Creutzfeld-Jakob disease was also included in the draft statement.
steam sterilization for instruments that have been used on patients known or suspected of having Creutzfeld-Jakob disease was also included in the draft statement.
Dry Heat
As mentioned previously, microbial inactivation due to exposure to dry heat is accomplished primarily as an oxidative process (3). Dry-heat sterilizers operate at atmospheric pressure, and although it is a relatively inefficient heat transfer agent, the fluid medium through which heat is dispersed to the surfaces of the load is air (3). Heat is transferred through air movement known as convection. Dry-heat sterilizers relying on natural convection due to temperature gradients in the chamber are the gravity convection type, while other, more efficient, equipment with fans to help with air movement are the mechanical convection or “forced-air” type of dry-heat sterilizers (3).
Dry-heat cycles are characterized by high heat, relatively long exposure periods, and sometimes lengthy lag times (especially in the gravity convection type equipment) as the load items are brought up to temperature. One factor that will impact the effectiveness of a dry heat process is the amount of water (intrinsic moisture) in the materials undergoing sterilization. The lower the RH, the longer the time required for sterilization using dry heat (23). Table 81-1 lists current time-temperature relationships for a variety of dry-heat sterilization cycles.
Dry-heat sterilizers should not be overloaded since air must freely circulate amongst the items in the load in order for proper heat transfer to occur. Dry heat is typically used to sterilize those items that would be damaged by steam and/or moisture or are otherwise impenetrable by steam. Examples of such items are petrolatum, oils, powders, delicate sharp instruments, and glassware (3). One useful application in particular is the use of dry heat to render glassware free of endotoxin residues during sterilization since cycles in steam autoclaves do not inactivate endotoxins. An additional benefit to using dry heat is its reduced propensity to corrode materials and dull sharp edges. Some of the obvious disadvantages, however, are (a) high temperature damage to easily charred or combustible materials, (b) long exposure times, (c) damage to some materials due to oxidation, and (d) a potential for distinct temperature zones being generated in the chamber due to improper loading and subsequent inefficient air circulation (3).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


