Figure 5-1. Horizontal Laminar Flow Workbench
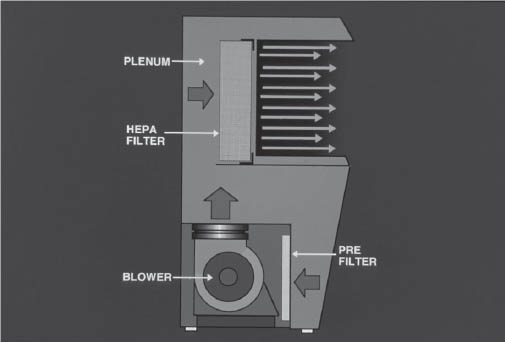
Illustration courtesy of the University of Tennessee Parenteral Medications Lab.
The plenum of the hood is the space between the prefilter and the HEPA filter. Air is pressurized here and distributed over the HEPA filter.
Laminar flow is the air in a confined space moving with uniform velocity along parallel lines. The term unidirectional flow has taken the place of laminar flow in more recent publications. Unidirectional flow is airflow moving in a single direction in a robust and uniform manner and at sufficient speed to sweep particles away from the critical processing area. Inside the HLFW is an ISO class 5 area (class 100 area).
Vertical Laminar Flow Workbench
The vertical laminar flow workbench (VLFW) works like an HLFW in that the air is drawn in through the prefilter and is pressurized in the plenum for distribution over the HEPA filter. However, the air is blown down from the top of the workstation onto the work surface, not across it (Figure 5-2).
Working in vertical laminar flow requires different techniques than does working in horizontal laminar flow. In vertical laminar flow, an object or the hands of the operator must not be above an object in the hood. In horizontal laminar flow, an object or the hands of the operator must not be in back of another object. The hands of the operator must never come between the HEPA filter and the object.
A biological safety cabinet, CAI, and CACI also use vertical unidirectional airflow to provide an ISO class 5 environment and are other examples of PECs.
Figure 5-2. Vertical Laminar Flow Workbench
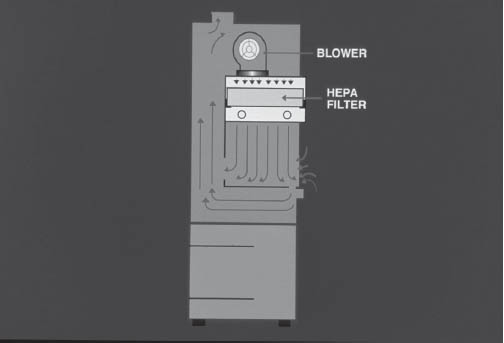
Illustration courtesy of the University of Tennessee Parenteral Medications Lab.
The HEPA Filter
The HEPA filter consists of a bank of filter media separated by corrugated pleats of aluminum. These pleats act as baffles to direct the air into laminar sheets. The HEPA filter is 99.97% efficient at removing particles 0.3 micron in size. Particles that are smaller or larger than 0.3 micron will be removed more efficiently.
Certification of the HEPA filter
The velocity of air from the HEPA filter is checked with a velometer or hot wire anemometer. ISO 14644 recommends that the average air velocity should be greater than 0.2 m/second.
Integrity of the HEPA filter: The dioctyl phthalate (DOP) test
The integrity of the HEPA filter is checked by introducing a high concentration of aerosolized Emery 3004 (a synthetic hydrocarbon) upstream of the filter on a continuous basis, while monitoring the penetration on the downstream side of the HEPA filter.
The aerosol has an average particle size of 0.3 micron. An aerosol photometer is used to check for leaks by passing the wand slowly over the filter and the gasket. None of the surfaces shall yield greater than 0.01% of the upstream smoke concentration. Any value greater than 0.01% indicates that a serious leak is present and must be sealed. All repaired areas must be retested for compliance. At one time, DOP was used to generate the aerosol. However, because DOP is a carcinogen, Emery 3004 is now used. An electronic particle counter cannot be used to certify the integrity of the HEPA filter. The particle counter is used to determine room classification.
Buffer Area (Controlled Area)
The PEC provides an ISO class 5 (class 100) area. It must be located in a controlled environment, away from excess traffic, doors, air vents, or anything that could produce air currents greater than the velocity of the airflow from the HEPA filter. Air currents greater than the velocity of the airflow from the HEPA filter may introduce contaminants into the hood. It is very easy to overcome air flowing at 90 feet per minute.
The buffer area shall be enclosed from other pharmacy operations. Floors, walls, ceiling, shelving, counters, and cabinets of the controlled area must be of nonshedding, smooth, and nonporous material to allow for easy cleaning and disinfecting. All surfaces shall be resistant to sanitizing agents. Cracks, crevices, and seams shall be avoided, as should ledges or other places that could collect dust. The floor of the buffer area shall be smooth and seamless with coved edges up the walls.
The walls of the buffer area can be sealed panels caulked with silicone or, if drywall is used, painted with epoxy paint, which is nonshedding. The corners of the ceiling and the walls shall be sealed to avoid cracks. A solid ceiling may be painted with epoxy paint, or nonshedding washable ceiling tiles that are caulked into place may be used.
Light fixtures shall be mounted flush with the ceiling and sealed. Anything that penetrates the ceiling or walls shall be sealed.
Air entering the room shall be fresh, HEPA filtered, and air conditioned. The room must be maintained in positive pressure (0.02–0.05 inch of water column) in relation to the adjoining rooms or corridors. If the buffer area is used for compounding of cytotoxic drugs, 0.01 inch of water column negative pressure is required. At least 30 air changes per hour shall occur, with the PECs allowed to provide up to 15 of the 30 required air changes per hour.
People entering the buffer area shall be properly scrubbed and gowned. Access to the buffer area shall be restricted to qualified personnel only.
Controlling the traffic in the buffer area is a critical factor in keeping the area clean. Only items required for compounding shall be brought into the buffer area. These items must be cleaned and sanitized before being taken into the buffer area. Items may be stored in the buffer area for a limited time. However, the number of items stored in the buffer area shall be kept to a minimum. All equipment used in the buffer area should remain in the room except during calibration or repair.
Because they can harbor many organisms, refrigerators and freezers should be located out of the buffer area. Computers and printers should be located outside of the buffer area because they generate many particles. However, if they are required to be in the buffer area, monitor the environment and evaluate their effect on the environment. Cardboard boxes shall not be stored in the buffer area. The items shall be removed from the boxes on the dirty side of the antearea and sanitized and transferred to the clean side of the antearea or to the buffer area for storage. Vials stored in laminated cardboard may be stored in the buffer area. Sinks or floor drains shall not be in the buffer area because potable water contains many organisms and endotoxins.
Preparation of Operators
An operator must be trained and evaluated to be capable of properly scrubbing and garbing before entering the buffer area. This requirement is critical to the maintenance of asepsis. The greatest source of contamination in a clean room is the people in the area. A seated or standing person without movement releases an average of 100,000 particles greater than 0.3 micron in diameter per minute. A person standing with full body movement releases an average of 2,000,000 particles per minute greater than 0.3 micron in diameter, and if moving at a slow walk, he or she releases an average of 5,000,000 particles. The garb is designed to help contain the particles that are being shed.
Before entering the antearea, an operator must remove all cosmetics and all hand, wrist, and other visible jewelry or piercings. Artificial nails or extenders are prohibited while working in the sterile compounding environment, and natural nails must be kept neat and trimmed. Garb is donned in an order proceeding from that considered dirtiest to that considered cleanest. Shoe covers, head and facial hair covers, and facemask or eye shields are donned before performing hand hygiene. Hands and forearms are then washed for 30 seconds with soap and water in the antearea, and hands and forearms are dried using a lint-free disposable towel or an electric hand dryer. While still in the antearea, an operator must don a nonshedding gown that zips or buttons up to the neck, falls below the knees, and has sleeves that fit snugly around the wrists. After entering the buffer area, an operator must use a waterless alcohol-based surgical hand scrub with persistent activity to again cleanse the hands before putting on sterile gloves. Sterile contact agar plates must be used to sample the gloved fingertips of compounding personnel after garbing to assess garbing competency. For successful completion of this competency, no colony-forming units can be found on any of the agar plate samples. Three consecutive, successful garbing and gloving exercises must be completed before sterile compounding is allowed. Routine application of sterile 70% isopropyl alcohol (IPA) must occur throughout the compounding process and whenever nonsterile surfaces are touched. After this initial evaluation, the entire process is repeated at least once a year for low- and medium-risk compounding and semiannually for high-risk compounding during any media-fill test procedure. The colony-forming unit action level for gloved hands will be based on the total number of colony-forming units on both gloves, not per hand.
Validation of the Operator
A media fill or media transfer is when a growth promotion media is used instead of the drug product, and all the normal compounding manipulations are done. It is critical that the process mimics the actual compounding process as closely as possible and represents worst-case conditions. Usually, the medium used is soybean-casein digest, which is also known as trypticase soy broth (TSB). This medium will support the growth of organisms that are likely to be transmitted to CSPs from the compounding personnel and environment. A media fill is used to check the quality of the compounding personnel’s aseptic technique. It is also used to verify that the compounding process and the compounding environment are capable of producing sterile preparations.
Initially, before an operator can compound low- or medium-risk sterile injectable products, he or she must successfully complete one media fill using sterile fluid culture media such as 3% soybean-casein digest medium. Media fill units must be incubated at 20–25°C for a minimum of 14 days or at 20–25°C for a minimum of 7 days and then at 30–35°C for a minimum of 7 days. A successful media fill is indicated by no growth in any of the media fill units. The media fill shall closely simulate the most challenging or stressful conditions encountered during the compounding of low- and medium-risk preparations. The compounding personnel shall perform a revalidation at a minimum of once a year by successfully completing one media fill. The media fills shall be designed to mimic the most challenging techniques the operator will use during a normal day. Validation for high-risk compounding focuses on ensuring that both the process and the compounding personnel are capable of producing a sterile preparation with all its purported quality attributes. Revalidation must be done on at least a semiannual basis. An example of a high-risk operation is the compounding of a sterile preparation from nonsterile drug powder. To mimic this operation, the compounder must use commercially available soybean-casein digest medium made up to a 3% concentration and perform normal processing steps, including filter sterilization. All media fills must occur in an ISO class 5 environment and must be completed without interruption.
5-6. Working in the Laminar Flow Workbench
Items not in a protective overwrap shall be wiped with a lint-free wipe soaked with sterile 70% IPA before being placed in the hood. Containers and packages must be inspected for cracks, tears, or particles as they are decontaminated and placed in the hood. Items in a protective overwrap, such as bags, must be taken from the overwrap at the edge of the hood (within the first 6 inches of the hood) and placed in the hood with the injection port facing the HEPA filter. The overwrap should not be placed in the hood, because doing so would introduce particles and organisms into the hood.
When working in the HLFW, an operator shall arrange supplies to the left or right of the direct compounding area (DCA). The critical site must be in uninterrupted unidirectional airflow at all times. The compounder must be careful not to place an object or hand between the HEPA filter and the critical site because doing so would interrupt the airflow to the critical site and potentially cause particles to be washed from the hand or object onto the critical site.
All work performed in the HLFW must be done at least 6 inches inside the hood. The unidirectional airflow is blowing toward the operator, who acts as a barrier to the airflow, causing it to pass around the body and create backflow. This turbulence can cause room air to be carried into the front of the hood.
Items placed in the HLFW disturb the unidirectional airflow. The unidirectional airflow is disturbed downstream of the item for approximately three times the diameter of the object. If the item is placed next to the sidewall of the hood, the unidirectional airflow is disturbed downstream of the item for approximately six times the diameter of the object. The area downstream from the nonsterile object is no longer bathed in unidirectional airflow and may become contaminated with particles. For these reasons, it is very important that a direct path exists between the HEPA filter and the area where the manipulations will occur.
With the VLFW, supplies in the hood should be placed so that the operator may work without placing a hand or object above the critical site. An operator can place many more items in the VLFW and still work without compromising the unidirectional airflow. Remember that within 1 inch of the work surface the air is turbulent. The unidirectional air, which is coming down from the HEPA filter, strikes the work surface and changes direction to move horizontally across the work surface. Therefore, all work in the VLFW should be done at least 1 inch above the work surface. During the compounding of sterile preparations, all movements into and out of the hood must be minimized to decrease the risk of carrying contaminants into the DCA. This can be achieved by introducing all items needed for the aseptic manipulation into the work area at one time and by waiting until the procedure is completed before removing used syringes, vials, and other supplies from the PEC.
5-7. Syringes, Needles, Ampuls, and Vials
Syringes
The basic parts of the syringe are the barrel, plunger, collar, rubber tip of the plunger, and tip of the syringe. Syringes are sterile and free of pyrogens. They are packaged either in paper or in a rigid plastic container. Syringe packages must be inspected to ensure that the wrap is intact and the syringe is still sterile. Syringes have either a Luer-Lok tip, in which the needle is screwed tightly onto the threaded tip, or a slip tip, in which the needle is held on by friction (Figure 5-3). Syringes are supplied with and without needles attached and are available in a variety of sizes. When removing the syringe from its package, take care not to let the syringe tip touch the surface of the hood.

Illustration courtesy of the University of Tennessee Parenteral Medications Lab.
Calibration marks are on the barrel of the syringe. These marks are accurate to one-half the interval marked on the syringe. The critical sites on the syringe are the tip of the syringe and the ribs of the plunger. The ribs of the plunger go back inside the syringe on injection of the fluid from the syringe and could potentially contaminate the syringe.
Needles
The basic parts of a needle include the hub, needle shaft, bevel, bevel heel, and tip of the needle (Figure 5-4).
Needles are sterile and are wrapped either in plastic with a twist-off top or in paper. This wrap must be inspected for integrity before the needle is used. The gauge of the needle refers to its outer diameter. The larger the number, the smaller the bore of the needle. The smallest is 27 gauge, and the largest is 13 gauge. The length of the needle is measured in inches, and some common lengths are 1–1.5 inches.
The critical sites on the needle are the hub of the needle, the entire needle shaft, and the tip of the needle.
Ampuls
Ampuls are single-dose containers. Once ampuls are broken, they are an open-system container; air can pass freely in and out of the ampul. Any solution taken from an ampul must be filtered with a 5 micron filter needle or filter straw, because glass particles fall into the ampul when it is broken. Before breaking the ampul, wipe the neck of the ampul with a sterile 70% IPA prep pad.
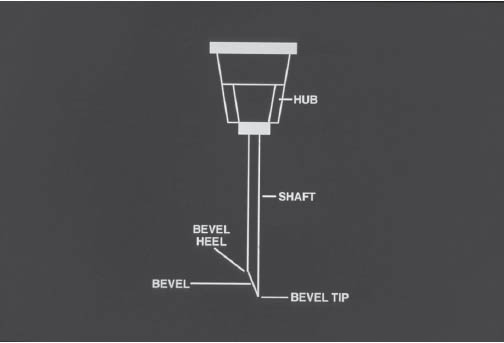
Illustration courtesy of the University of Tennessee Parenteral Medications Lab.
Vials
A vial is a molded glass or plastic container with a rubber closure secured in place with an aluminum seal. It may contain sterile solutions, dry-filled powders, or lyophilized drugs, or it may be an empty evacuated container. Vials may be single-dose or multiple-dose containers.
A single-dose container usually contains no preservative system to prevent the growth of microorganisms if they are accidentally introduced into the container. A single-dose vial punctured in an environment worse than ISO class 5 air must be used within 1 hour. A single-dose vial continuously exposed to ISO class 5 air may be used up to 6 hours after initial needle puncture. When the vial is first used, it should be labeled with the date, time, and initials of the person using the vial so the beyond-use date (BUD) can be determined. A multidose vial contains preservatives, and these vials can be entered more than once. The pharmaceutical manufacturer has done studies to prove that the preservative system will remain effective and the closure will reseal after penetration by the needle. Therefore, the BUD for opened or entered multidose containers is 28 days, unless otherwise specified by the manufacturer.
5-8. Biological Safety Cabinets
A class II biological safety cabinet (BSC) should be used to prepare cytotoxic and other hazardous drugs. Four different types of class II BSCs exist. Types A1 and A2 exhaust 30% of HEPA-filtered air either into the room or to the outside through a canopy connection. Type A1 mixes the supply air in a common plenum and may have ducts and plenum under positive pressure. Type A2 has all contaminated ducts and plenum under negative pressure or surrounded by negative pressure. Type B1 exhausts 70% of total air through a dedicated exhaust duct and must be hard ducted. Type B2 exhausts 100% of total air to the outside without any recirculation and must be hard ducted also. With types B1 and B2, all the ducts and the plenum are under negative pressure and are surrounded by negative pressure.
Preparation of Hazardous Drugs
When working with hazardous drugs, personnel must wear appropriate protective equipment, including solid-front gowns, face masks, eye protection, hair and shoe covers, and double sterile chemotherapy-type gloves. Personnel must handle all hazardous drugs with caution at all times, using appropriate chemotherapy gloves, not only during preparation, but also during receiving, distribution, stocking, inventorying, and disposal.
It is imperative that positive pressure not be allowed to build up in the vial. Proper training on the use of a chemotherapy-venting device, which uses a 0.2 micron hydrophobic filter or the negative pressure technique to prevent the build up of positive pressure within the vial, must be done before preparing hazardous drugs and on an annual basis. When a closed system transfer device (one that allows no venting or exposure of hazardous substance to the environment) is used, it shall be used within the ISO class 5 environment of a BSC or CACI.
When compounding, personnel must use syringes and IV sets with Luer-Lok fittings if possible. Use a large enough syringe so that the plunger does not separate from the barrel of the syringe when filled with solution. Syringes should be filled with no more than 75% of their total volume. When possible, attach IV sets and prime them before adding the hazardous drug. Wipe the outside of the bag or bottle to remove any inadvertent contamination. The use of nonshedding plastic-backed absorbent pads is also conducive to keeping the BSC as clean as possible.
The PEC shall be located in an ISO class 7 area physically separated from other preparation areas and maintained under negative pressure of not less than 0.01 inch water column to the surrounding area.
5-9. Overview of the Standard of Practice Related to Sterile Preparations: The United States Pharmacopeia (USP) 37/National Formulary (NF) 32
Chapter <797>, Pharmaceutical Compounding—Sterile Preparations in USP 32/NF 27, became the official standard for sterile pharmaceutical compounding in June 2008. Chapter <797> has three microbial risk levels of compounded sterile preparations. The risk levels are determined on the basis of the potential for the introduction of microbial, chemical, or physical contamination into the product. The chapter covers topics such as validation of sterilization and of the aseptic process, environmental control and sampling, end-product testing, bacterial endotoxins, training, and a quality assurance program.
Low-Risk Compounding
Compounding is classified as low risk when all of the following conditions prevail:
■ Commercially available sterile products, components, and devices are used in compounding within air quality of ISO class 5 or better.
■ Compounding involves few aseptic manipulations, using not more than three commercially manufactured sterile products and not more than two entries into any one sterile container.
■ Closed-system transfers are used. Withdrawal from an open ampul is classified as a closed system.
■ In the absence of passage of a sterility test, the storage periods for the CSPs cannot exceed the following time periods before administration:
• Storage for not more than 48 hours at a controlled room temperature of 20–25°C
• Storage for not more than 14 days at a cold temperature of 2–8°C
• Storage for not more than 45 days in a solid frozen state between –25°C and –10°C.
Medium-Risk Compounding
Medium-risk CSPs are those compounded under low-risk conditions when one or more of the following conditions exist:
■ Compounding involves pooling of additives for the administration to either multiple patients or to one patient on multiple occasions.
■ Compounding involves complex manipulations other than a single volume transfer.
■ The compounding process requires a long time period to complete dissolution or homogeneous mixing.
■ In the absence of passage of a sterility test, the storage periods for the CSPs cannot exceed the following time periods before administration:
• Exposure for not more than 30 hours at a controlled room temperature of 20–25°C
• Storage for not more than 9 days at a cold temperature of 2–8°C
• Storage for not more than 45 days in a solid frozen state between –25°C and –10°C.
High-Risk Compounding
High-risk compounds are compounded under any of the following conditions and are either contaminated or at high risk to become contaminated with infectious microorganisms:
■ A sterile preparation is compounded from non-sterile ingredients.
■ Sterile ingredients or components are exposed to air quality inferior to ISO class 5 for more than 1 hour, including storage in environments inferior to ISO class 5 of opened or partially used packages of manufactured sterile products with no antimicrobial preservative system.
■ Nonsterile water–containing preparations are exposed for more than 6 hours before being sterilized.
■ No examination of labeling and documentation from suppliers or direct determination that the chemical purity and content strength of ingredients meet their original or compendia specification occurs.
■ Compounding personnel are improperly garbed and gloved.
■ In the absence of passage of a sterility test, the storage periods for the CSPs cannot exceed the following time periods before administration:
• Storage for not more than 24 hours at a controlled room temperature of 20–25°C
• Storage for not more than 3 days at a cold temperature of 2–8°C
• Storage for not more than 45 days in a solid frozen state between –25°C and –10°C.
5-10. Sterilization Methods
Filtration
Filtration works by a combination of sieving, adsorption, and entrapment. Care must be taken to choose the correct filter to sterilize the preparation. Membrane filters generally are compatible with most pharmaceutical solutions, but interactions do occur—often because of sorption or leaching. Sorption is the binding of drug or other formulation components to the filter, which can occur with peptide or protein formulations. There are filters that have little or no affinity for peptides or proteins. Leaching is the extracting of components of the filter into the solution. Surfactants are often added to the filter to make it hydrophilic, and they may leach into the product. Large-molecular-weight peptides may be affected by filtration. Their passage through a filter with a small pore size may cause shear stress and alter the three dimensional structure of the peptide. Solvents in the parenteral formulation may also affect filters. All filter manufacturers have compatibility data on their membrane type and can be a great source of information when choosing a membrane.
Filter choice
Choose the appropriate size and configuration of filtration device to accommodate the volume being filtered and permit complete filtration without clogging of the membrane. A 25 mm syringe disk filter should filter no more than 100 mL of solution. If the solution being filtered has a heavy particulate load, a 5 micron filter should be used before the 0.2 micron filter to decrease the particulate load to the 0.2 micron filter. The filter membrane and housing must be physically and chemically compatible with the product to be filtered and capable of withstanding the temperature, pressures, and hydrostatic stress imposed on the system.
A pharmacy may rely on the certificate of quality provided by the vendor. Certification shall include microbial retention testing with Brevundimonas diminuta at a minimum concentration of 107 organisms per cm2, as well as testing for membrane and housing integrity, nonpyrogenicity, and extractables.
Hydrophobic and hydrophilic filters
Hydrophilic membranes wet spontaneously with water. They are used for filtration of aqueous solutions and aqueous solutions containing water-miscible solvents. Hydrophobic filters do not wet spontaneously with water. They are used for filtering gases and solvents.
Filter integrity
A sterilizing filter assembly shall be tested for integrity after filtration has occurred. The bubble point
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


