Commercial Support Stability
As part of the submission process, a company will include a commitment to continue stability evaluation over the commercial life of the product. The stability commitment may vary depending on the properties of the product, but generally specifies that one representative batch per year be held at the long-term storage condition (Table 25.1) and tested at the same time points and by the same methodology as used to generate the submission data.
The results generated for a commitment batch have far-reaching implications. Thus, a batch that fails to meet specification during shelf life would cause all batches of the same packaging configuration to be evaluated. In such a scenario, a firm could be required to reduce expiry or recall product from the market.
Change Management Stability
The components, processing, packaging, and labeling of a drug product are specified in the approved application. A change to any of these requires evaluation to ensure that the change has no deleterious effect on the product. Stability testing is frequently a part of this evaluation. For example, a firm may wish to change the supplier of the active pharmaceutical ingredient (API) in their drug product. This change would require stability testing. Just as in the original application, the results will be evaluated and submitted to regulatory authorities for approval. The magnitude of change and the tolerances established during registration will influence the type of stability program required to support the change.
Execution of Stability Studies
As with any good scientific evaluation, stability studies begin with outlining the steps to be taken and the rules for interpreting the results (that is, this plan is the stability protocol). Minimally, a protocol should specify the sample storage conditions/testing time points/packaging configuration and provide reference to the test methods/acceptance criteria against which the test article is being evaluated. Studies performed to support a regulatory submission generally include the criteria for evaluating the data.
Conditions
The ICH Q1A (R2) guidance outlines the generally accepted conditions at which studies are to be performed (Table 25.1). Exceptions to these conditions, with a written justification, can be made depending on the nature of the product and the intended use of the data.
Time Points
The Q1A guidance provides recommended time points for testing. These are typically one, two, three, and six months for accelerated studies; and three, six, nine, 12, 18, 24, and 36 months for long-term studies. The intermediate storage condition is set up to provide additional information should a study fail to meet acceptance criteria during the accelerated study. Like the storage conditions, these may be adjusted as needed to fulfill the purpose of the study.
Packaging Configurations
Each unique presentation of a product must be represented in the commercial support stability program. For example, a drug product that has one strength and is in four package configurations would have four unique presentations of the product. Thus, the four presentations need be represented on stability.
A stability program can be established by bracketing. Bracketing is based on the extremes, which are put on stability, and the intermediates are assumed to be represented. For example, bracketing can be applied to different container sizes that are made from the same resin, filled with the same product. If a drug product is packaged in the same bottle with three different tablet counts (30-count, 90-count, and 100-count) and in a unit-dose blister, only three presentations (30-count, 100-count, and unit-dose blister) will be on stability. The fourth configuration (90-count), which is bracketed by the 30-count and 100-count, is assumed to be represented. Matrixing can be used. With matrixing, a selected subset of the total number of possible samples with all factor combinations is tested at a specified time point.
Once data are generated from the study, they will need to be evaluated. The first step in this process is to compare them against the acceptance criteria. The second step is to look at the trending. Trending is typically performed on sets of data that include the same formulation and packaging configuration. An approach for analyzing stability data, which is expected to change over time, is to determine the time at which the 95% one-sided confidence limit for the mean curve intersects the acceptance criterion (Figure 25.1).
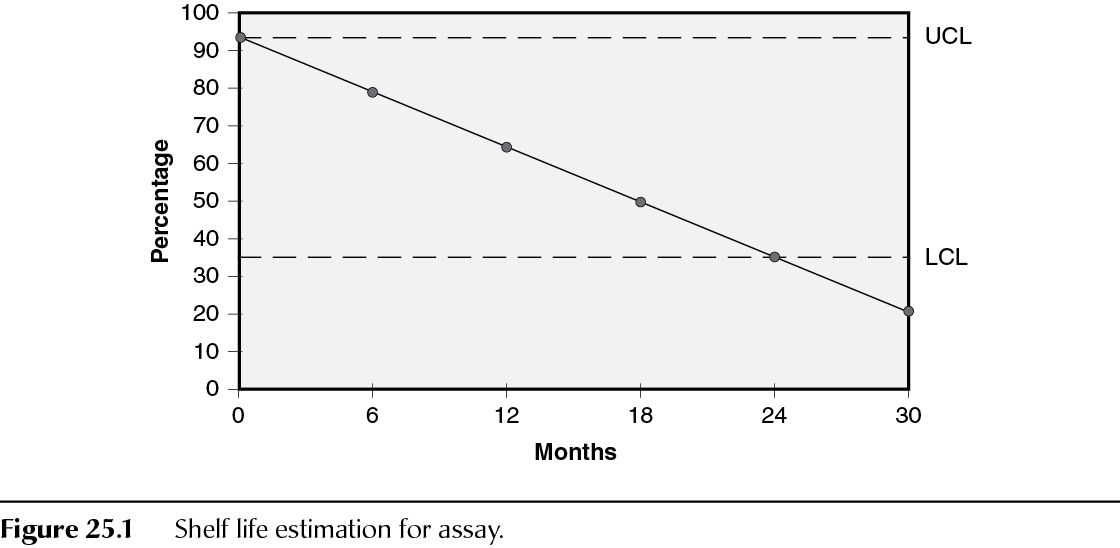
Based on the stability data extrapolation, companies can infer information about future data and establish dating. This information can aid in establishing acceptance criteria/expiry dating for the new product based on a high degree of confidence. After approval of a new material, the ongoing stability study data will permit the detection of a stability issue, such as change in impurity levels. In general, certain quantitative attributes, such as assay and degradation products, can be assumed to be zero-order kinetics during long-term storage, thus the trending and regression statistics can be applied to these stability-indicating tests. Although the kinetics of other quantitative attributes, such as pH and dissolution, are not known, the same statistical analysis model can be applied.
Results that do not meet the acceptance criteria or demonstrate adverse trending must be investigated both from a laboratory perspective (was the testing performed correctly?) and from a product perspective (were formulation, manufacturing, packaging, and delivery performed correctly?). Typically, an out-of-specification (OOS) procedure will be followed to conduct the investigation. For approved products, and if the stability study is representative of product in commerce, a confirmed OOS result will require notification to the United States Food and Drug Administration (FDA) within three days of the confirmation. Depending on the failure and product evaluation, a company may potentially reduce the expiry dating or recall the product.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


