Table 9.2 ICH climatic zones and their associated long-term storage conditions.
| Climatic zone | Definition | Long-term storage conditions |
| I | Temperate | 21 °C, 45% RH |
| II | Subtropical and Mediterranean | 25 °C, 60% RH |
| III | Hot and dry | 30 °C, 35% RH |
| IVA | Hot and humid | 30 °C, 65% RH |
| IVB | Hot and very humid | 30 °C, 75% RH |
- Stability testing protocols are defined in ICH Guidelines Q1A(R2) and comprise long-term, intermediate, accelerated and stress conditions.
- Long-term storage can also mean refrigerated or frozen, depending on the stability of the drug substance.
- Storage conditions vary around the world, so four climatic zones are defined.
9.2 Degradation mechanisms
Chemical degradation usually occurs via one of three reaction types:
- Hydrolysis or solvolysis
- Oxidation
- Photolysis
The rates of all reaction types may be increased with a catalyst. In addition, physical changes may occur that affect product performance, such as a change of polymorph or loss/formation of a hydrate form. The primary outcomes of stress testing during preformulation should be to determine:
This information should be sufficient in order to make a go/no go decision in respect of each drug substance. Once the underlying degradation profile of a drug substance is understood, dosage form, excipient choice, processing conditions and packaging selection can be made to mitigate any stability issue. The rate of degradation in the solid-state usually varies between polymorphic or pseudopolymorphic forms. Stress testing in solution will identify primary degradation pathways, but solid-state stability testing of the physical form selected for development will be necessary in the medium-term. Long-term and accelerated stability programmes can then be designed and implemented to provide data for regulatory submission. Since physical form characterisation was discussed in Chapters 7 and 8, the text below considers chemical degradation.
9.2.1 Hydrolysis
Hydrolysis reactions involve chemical reaction between at least two species, one of which is water. Although hydrolysis reactions should follow second-order kinetics (see below), because water is present in excess the reaction rate will be experimentally measured as pseudo first-order, being dependent upon the concentration of solute only. Chemical groups that are likely to hydrolyse include esters, β-lactams, amides and some imines. Generally amide bonds hydrolyse more slowly than esters. This means amide drug substances may be more stable than their ester counterparts. For instance, procaine (an ester) hydrolyses during autoclaving while procainamide does not. Similarly, hydrolysis rates are hugely dependent upon chemical structure, reflecting the ease with which nucleophilic attack can occur or the stability of any intermediate structures; procaine has a short shelf life in solution (less than 2 days at 25 °C; Loucas et al., 1981), while lignocaine is remarkably stable with respect to temperature, acid and alkali.
In a review of the literature, Day and Ingold (1941) proposed a standard nomenclature for eight standard mechanisms of hydrolysis. The system uses four characters. The first character is either A or B, for acid or base hydrolysis, and the last character is either 1 or 2, for unimolecular or bimolecular. The middle two characters were originally prime or double prime but were revised by Ingold (1953) to AC or AL, representing scission of either the acyl or alkyl oxygen respectively. The two most common mechanisms are AAC2 and BAC2, reflecting the fact that the acyl bond is more easily broken than the alkyl bond and that bimolecular mechanisms are favoured over unimolecular mechanisms. Both AAC2 and BAC2 proceed via formation of a tetrahedral intermediate and it is thought that two molecules of water are involved (tetrahedral intermediates require three molecules to collide and interact, which is unusual, but in this case the two water molecules are already interacting through a hydrogen bond). For instance, in the case of AAC2,
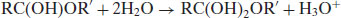
The AAL1 mechanism also proceeds very rapidly in cases where the R′ group can exist as a stable carbonium ion.
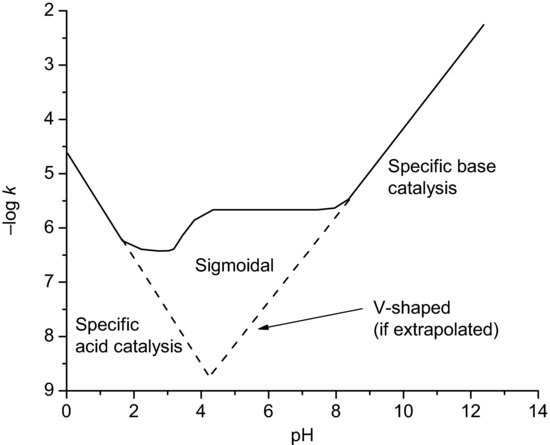
Where specific acid or base-catalysed hydrolysis occurs, the stability–pH profile of a drug substance will not be linear. The pH of maximum stability typically falls between 2.5 and 7, which reflects the fact that the hydroxyl ion attacking a carbonyl carbon (of an ester or amide) is a better nucleophile than water attacking a protonated carbonyl. Stability–pH profiles show various behaviours, depending upon the mechanism of reaction:
- V- or U-shaped. Indicative of specific acid catalysis at low pH and specific base catalysis at high pH. Figure 9.1 shows the V-shaped profile for atropine.
- Sigmoidal. Indicative of dissociation of the functional group undergoing nucleophilic attack. Figure 9.2 shows the sigmoidal profile for cycloserine.
- Bell-shaped. Indicative of dissociation of two functional groups. The overall profile is the sum of two mirror-image sigmoidal curves and maximum instability is seen at the point of intersection. Figure 9.3 shows the bell-shaped profile for aztrenam.
Figure 9.1 V-shaped pH–stability profile for hydrolysis of atropine (redrawn from Connors et al. (1986)).
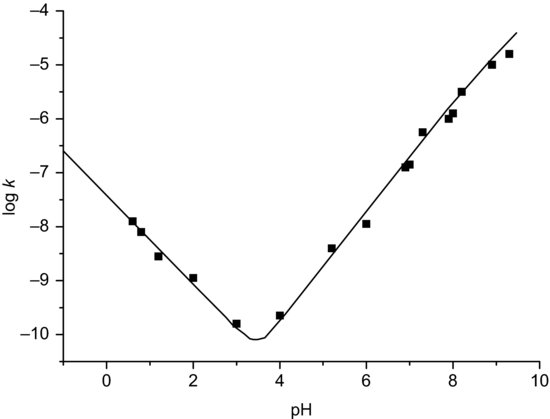
Figure 9.2 Sigmoidal pH-stability profile for hydrolysis of cycloserine in dilute solution (redrawn from Kondrat’eva et al. (1971), with kind permission from Springer+Business Media B.V.).
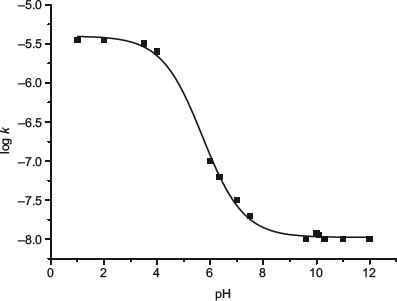
Figure 9.3 Double bell-shaped pH–stability profile for hydrolysis of aztreonam (redrawn from Connors et al. (1986)).
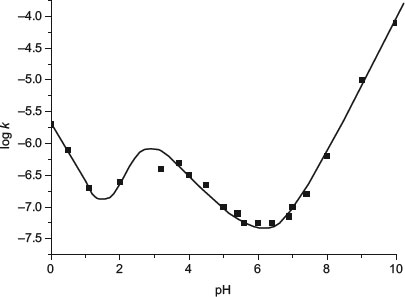
The experimentally observed profile may comprise more than one of these behaviours. For instance, the profile for aspirin (Figure 9.4) shows V-shaped behaviour at the extremes of pH, with sigmoidal behaviour in between.
Figure 9.4 pH profile for the hydrolysis of aspirin, showing V-shaped behaviour at extremes of pH and sigmoidal behaviour in between (redrawn from Connors et al. (1986)).
Study question 9.1 It seems sensible to formulate a drug substance at the pH of maximum stability. Why might this be a problem?
9.2.2 Solvolysis
Solvolysis is the same process as hydrolysis but the reactant is a solvent other than water (for instance, reaction with methanol is methanolysis, reaction with ethanol is ethanolysis, etc.).
Drug substances are generally more stable in organic solvents than in water, although photolysis and oxidation reactions may proceed at a faster rate as free radicals are usually more stable in less polar solvents. Thus, solvent polarity rather than pH is the greatest factor affecting degradation rates. As a general rule if the degradation products are more polar than the parent molecule then addition of a less polar solvent will increase stability. Similarly, where the degradation products are less polar than the parent molecule then addition of a more polar solvent will increase stability. If the drug substance and any degradants are nonpolar, such as the steroids, then there will be no change in stability with polarity and so any (physiologically acceptable) solvent may be added to increase solubility without affecting stability.
9.2.3 Oxidation
Oxidation originally meant reaction of a compound with oxygen to produce an oxide, but is now recognised as any reaction where the oxidation state of the reacting molecule is increased (in other words, it loses at least one electron). By definition therefore the oxidising agent must itself gain at least one electron and is reduced, forming a free radical. Oxidation reactions are thus said to involve a redox pair. Typically, oxidising agents are compounds that contain elements in high oxidation states (such as H2O2, MnO–4 or Cr2O2-7) or highly electronegative atoms that can easily accommodate an extra electron (such as O2, Fe2 or Cl2). From a stability perspective, reaction with oxygen is of the greatest interest, since it is present in the environment while other oxidising agents would have to have been specifically added to the formulation, although metal ions may be present in solutions to an appreciable degree and these often catalyse oxidative processes. One method to check whether metal ions are catalysing oxidation is to add a metal chelator (such as ethylenediamine tetra-acetic acid, ETDA) or antioxidant to the solution; this will bind any metal ions, reducing oxidation of the drug substance (Figure 9.5).
Figure 9.5 The effect of various antioxidants, chelating agents and antioxidant/chelating agent mixtures on the formation of peroxides in oil of terebinth (redrawn from Fryklöf (1954)).
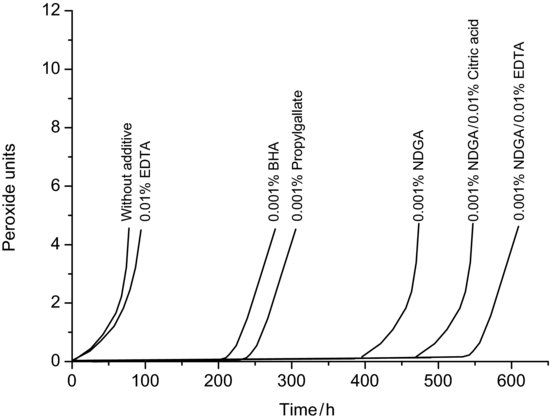
When a drug substance reacts with atmospheric oxygen spontaneously at room temperature the process is known as auto-oxidation. Molecular structures that tend to oxidise include heterocyclic aromatic rings, those with a hydroxyl group bonded directly to an aromatic ring, conjugated dienes, nitroso and nitrate derivatives and aldehydes. Examples of drugs that are susceptible to auto-oxidation include adrenaline, ascorbic acid, heparin, hydrocortisone, morphine, the penicillins, the tetracyclines and some of the oils used in flavouring. One outcome from this is that as well as loss of potency, the drug product may also start to taste unpleasant with time. It is also typical that degradants of oxidation are coloured. Even when the amount of degradation is within tolerance, the degradants may discolour the product.
Study question 9.2 How might oxidative degradation be reduced and, if some oxidation occurs during storage, how might discolouration be masked in the drug product?
9.2.4 Photolysis
Photolysis means reaction of the drug substance or product with light. Demonstration of the photostability of a new drug substance is a requirement of any development programme and is covered by ICH Guideline Q1B (1996). The intrinsic photostability characteristics of a drug substance should be characterised so that exposure to light does not result in an unacceptable change. During preformulation, photostability testing may be performed on a single batch of drug substance under stress conditions. If instability is seen, then confirmatory studies must be performed under standard conditions. Two light sources are specified:
- Any light source that produces an output similar to the D65/ID65 emission standards (these are the outdoor daylight (D65) and indoor daylight (ID65) standards defined in ISO 10977, 1993). Typical examples of such light sources include fluorescent lamps with visible and UV outputs and xenon or metal halide lamps. Significant light emission below 320 nm may be removed with a filter.
- A combination of a cool white fluorescent lamp (producing an output similar to that specified in ISO 10977, 1993) and a near-UV fluorescent lamp with a spectral distribution between 320 and 400 nm and with a maximum output between 350 and 370 nm.
Light is a form of energy, propagated as an electromagnetic wave, and so irradiation of a drug substance with light can potentially initiate several processes. Energy may be absorbed by a drug substance, transferred to other molecules or emitted at a different frequency, resulting in possible degradation and/or an increase in temperature. Light energy may also promote oxidation and hydrolysis, so photostability testing in solution is as important as photostability testing in the solid-state. Since light energy is inversely proportional to wavelength, UV frequencies generally cause more degradation than visible frequencies.
Photolysis can involve multiple mechanisms and reaction pathways and a complex mixture of degradants can be created; the exact composition will be dependent upon the spectral distribution of the light source. Where maximum degradation is seen to occur at a specific frequency of light (the causative wavelength) packaging can be designed to reduce exposure of the drug substance to that wavelength. Plain glass absorbs more than 80% in the 290–320 nm region, while amber glass increases absorption to nearly 95%. It is also possible to add UV blockers or a film coat with an opaque layer.
Where a drug is photolabile, degradation can occur very rapidly (nifedipine, for instance, has a half-life of only a few minutes). One point to note is that rapid degradation at a specific wavelength (photosensitivity) can be a formulation strategy, often used as a targeting strategy for chemotherapy agents.
- The principal causes of chemical degradation are hydrolysis (or solvolysis), oxidation and photolysis.
- Change in physical form may also reduce drug product performance.
- Hydrolysis is often catalysed in acidic or basic conditions. A stability–pH profile is useful in identifying a hydrolysis mechanism.
- Oxidation can lead to change in taste and/or discolouration as well as loss of drug substance.
- Catalysis generally acts to speed hydrolysis and oxidation.
9.3 Reaction kinetics
Most drug products are formulated as solids, and so solid-state reaction kinetics generally govern the shelf life of drug products. Initial formulations, for toxicology or bioavailability studies, however, may well be solutions, in which case solution-state kinetics predominate, or suspensions, in which case pseudo zero-order kinetics often predominate. In either event, understanding the principles of reaction kinetics will facilitate quantitative interpretation of stability data and indicate the potential reaction mechanism.
9.3.1 Solution-phase kinetics
Solution-phase kinetics are based on the concept of molecularity (i.e. on reaction stoichiometry). This posits that in solution reactants are free to move and so the reaction rate is proportional to the number of collisions between molecules. Consider the reaction scheme
(9.1)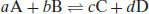
The reaction rate can be defined in terms of the forward reaction (the reacting species):
(9.2)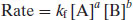
or in terms of the reverse reaction (the products):
(9.3)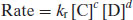
where the superscripts (a and b or c and d) sum to give the order of reaction and k is a constant of proportionality. At equilibrium the rates of the forward and reverse reactions must be equal and so
(9.4)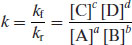
where k is termed the rate constant. Solution-phase reactions are typically zero, first or second order. Noninteger orders may be experimentally measured, but this indicates that degradation is progressing via multiple pathways.
9.3.2 Zero-order reactions
If the order of reaction is zero, then the reaction rate is independent of the concentration of any of the reacting species and is constant:
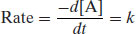
When integrated, Equation (9.5) becomes
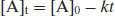
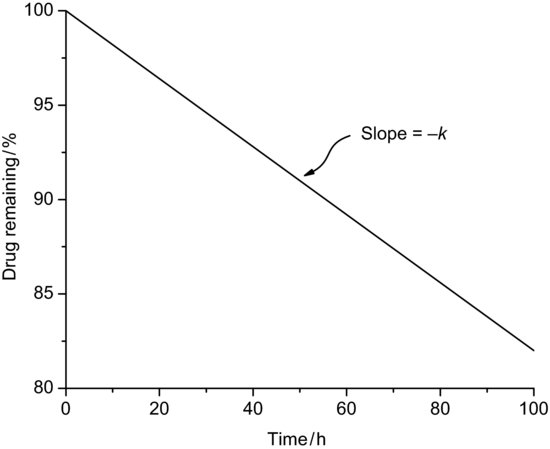
where [A]t is the concentration of reactant A at any time and [A]0 is the initial concentration of reactant A. Hence a plot of concentration versus time is linear with slope –k (Figure 9.6). There are very few truly zero-order reactions (if a reaction is seen to degrade with zero-order kinetics it is usually pseudo zero-order, principally because one of the reacting components is present in great excess – this is the case for suspensions noted earlier, where the solution concentration is kept constant by dissolution of material from the solid phase). Zero-order rate constants have units of concentration time−1.
< div class='tao-gold-member'>