Fig. 16.1.
Approach to examination of the spleen.
Grossly, normal spleen has small white flecks in a background of purple-red, representing the white and red pulp, respectively
The gross presentation of white pulp lesions is a caricature of the normal splenic appearance; the small white flecks expand in size and become readily visible small white nodules, which is described as a miliary pattern (Fig. 16.2)
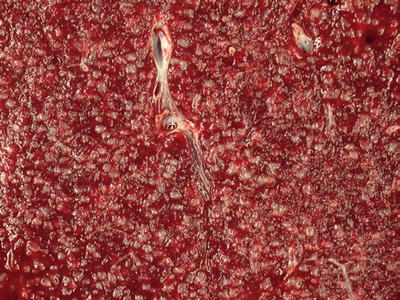
Fig. 16.2.
White pulp: the miliary pattern is an accentuation of the normal splenic architecture. The white pulp components are enlarged and prominent.
In red pulp lesions, the spleen takes on a beefy red appearance, with the white pulp becoming inapparent
Focal lesions can vary in size, consistency, and circumscription of their boundaries; these may represent cystic or solid masses. Color and density of mass lesions are dependent on their cellular composition, although many focal lesions in spleen evoke a fibrosclerotic response from the splenic stroma, imparting a firm, yellow tan appearance (Fig. 16.3)
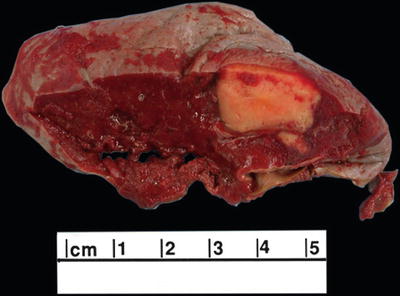
Fig. 16.3.
Splenic infarction: the firm, tan wedge-shaped mass is a splenic infarction. Microscopically, it is composed of macrophages, fibroblasts, and benign vascular elements.
♦
The spleen as an organ is unique in its combination of functions:
The spleen functions as part of both the immune system and the hematopoietic system
As an immune organ, one of its functions is to serve as a very large lymph node, with immunologic responses comparable to those found in other secondary lymphoid organs
Its immunologic function also includes phagocytosis of cells or materials that are recognized as “foreign”
In a similar manner, the spleen serves to cull effete red blood cells as they pass through splenic sinuses into cords; red blood cells lacking appropriate membrane deformability will be repaired or destroyed as they transit through the acidotic environment
The splenic architecture is composed of a fibrous capsule with an interconnected complex of support trabeculae, a network of arteries and arterioles, a complex arrangement of vascular sinuses with associated macrophages, and a complex arrangement of lymphoid tissue
Because of this mixed cellular composition and diverse functionality, the spleen presents with diseases that are expressions of its similarities to other sites (lymphomas) and its abundance of certain types of tissues (vascular tumors) but also related to its functions (extramedullary hematopoiesis, acute leukemias) and its unique anatomy (littoral cell angiomas)
Because of its filtering function, it is not uncommon for the spleen to be a site of infection. The microenvironment of the spleen is somewhat inhospitable, so the organisms that are present must be particularly good at evading immunologic destruction (e.g., encapsulated bacteria, fungi)
Splenomegaly
♦
Splenomegaly may result from a variety of causes, both benign and malignant. Some of the most common causes of splenomegaly are systemic disorders that merely represent hyperfunction of normal splenic activities, stromal proliferation, and/or vascular congestion
♦
Splenomegaly may result in clinical symptoms including early satiety, abdominal fullness, and pain
♦
Splenic enlargement is not an uncommon manifestation of systemic hematologic disease; however, the spleen is rarely removed as a diagnostic specimen. The spleen may be removed as a result of intractable symptoms, including patient discomfort or cytopenias . Only rarely is the spleen removed for evaluation of an unknown cause of splenomegaly or a mass of uncertain etiology
Splenic Rupture (Fig. 16.4)
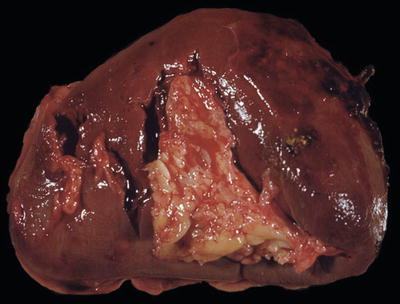
Fig. 16.4.
Splenic rupture: this gross photograph shows several ruptures in the splenic hilum and with small amounts of adherent blood clot.
♦
There are a wide variety of both nonneoplastic and neoplastic processes associated with splenic rupture. The spleen is typically enlarged in these cases, sometimes massively. The rapidity of enlargement may also play a role with acute processes rupturing while the spleen is smaller
♦
Hemoperitoneum as a result of splenic rupture often leads to serious consequences including shock and death, if not treated appropriately
♦
Trauma is the most common cause of splenic rupture. The so-called spontaneous splenic rupture is better termed “atraumatic” rupture , which has some underlying cause. Spontaneous splenic rupture should be reserved for cases where no etiology is identified. A list of causes associated with splenic rupture can be seen in Table 16.1. In cases with an underlying neoplasm, atraumatic splenic rupture is associated with a high mortality rate. One notable cause of atraumatic splenic rupture is infectious mononucleosis/acute Epstein–Barr virus infection
Table 16.1.
Atraumatic (“Spontaneous”) Splenic Rupture: Associations and Etiologies
I. Infectious |
a. Bacterial i. Typhoid fever ii. Q fever iii. Infectious endocarditis (various species) iv. Tuberculosis and other mycobacteria v. Numerous others b. Viral i. Epstein–Barr virus ii. CMV iii. HIV iv. Others c. Parasitic/protozoal i. Malaria ii. Others |
II. Hematopoietic diseases a. Acute leukemias b. Lymphoma c. Myeloma/plasma cell neoplasms/amyloidosis (esp. plasma cell leukemia) d. CML and other myeloproliferative neoplasms (PMF, ET, PV) |
III. Vascular lesions a. Benign (e.g., peliosis, hemangioma) b. Neoplastic (e.g., littoral cell angioma, angiosarcoma) |
IV. Cysts |
V. Infarction |
VI. Autoimmune disorders including collagen-vascular diseases |
VII. Hamartoma |
VIII. Medications a. Anticoagulants and thrombolytics b. Cytokines including G-/GM-CSF |
IX. Metastatic malignancy (carcinoma, melanoma, etc.) |
X. Storage diseases |
XI. Coagulation abnormalities a. Factor VIII deficiency b. Congenital dysfibrinogenemia |
XII. Pregnancy related a. Postpartum b. Ectopic pregnancy (“splenic” pregnancy) |
XIII. Miscellaneous a. Sarcoidosis b. Acute pancreatitis c. Cirrhosis of the liver from any cause d. Mechanical (after vomiting, coughing) |
Splenic Ectopia
♦
Accessory spleens are not uncommon findings at autopsy (20–30% of population) and are typically located in the splenic hilar region but can be located at more distant sites. Accessory spleens have the normal architecture of spleen and are typically small
♦
Splenic–gonadal fusion is a rare developmental anomaly with portions of the spleen attaching to the left gonad +/− fibrous tissue connecting the normally located spleen with the wandering fragment
♦
Splenosis refers to the presence of multiple small foci of splenic tissue, which are present in the abdominal cavity. Splenosis usually arises after rupture of the spleen and seeding of the abdominal cavity with small fragments of splenic parenchyma
Lymphoid Neoplasms
Lymphomas Presenting Predominantly in White Pulp
Splenic B-Cell Marginal Zone Lymphoma
Definition
♦
B-cell lymphoma, thought to be derived from splenic marginal zone cells
♦
Splenic involvement is predominant, although marrow and peripheral blood involvement are present in virtually all cases
♦
Formerly known as “splenic lymphoma with villous lymphocytes”
♦
Splenic B-cell marginal zone lymphoma (SMZL) is a relatively common diagnosis made in spleens removed for undetermined causes of splenomegaly
Clinical
♦
Patients most often present with asymptomatic splenomegaly
Laboratory
♦
Leukocytosis may be present. Lymphocytes in peripheral blood may have cytoplasmic projections, referred to as villous lymphocytes
♦
Spleens are typically enlarged, often massively, with prominent expansion of the white pulp in a miliary pattern
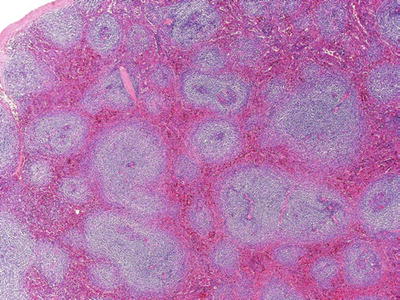
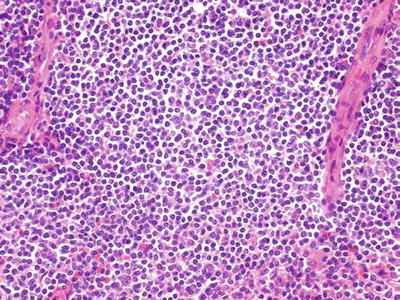
Fig. 16.5.
Splenic marginal zone lymphoma: expansion of the white pulp by pale-appearing lymphocytes.
Fig. 16.6.
Splenic marginal zone lymphoma: predominantly small lymphocytes with moderate amounts of clear or pale cytoplasm.
Microscopic
♦
The white pulp is expanded and replaced by a population of predominantly small lymphocytes. These are typically round, with a relative increase in pale cytoplasm giving them a pale appearance; rare larger, transformed lymphocytes are also present. Occasionally plasma cell differentiation can be identified. With early involvement, these lymphomas will expand the normally present marginal zone and be difficult to distinguish from marginal zone hyperplasia
♦
Another diagnostic feature is replacement of the periarteriolar lymphoid sheaths with B cells. As the disease progresses, they will invade and replace the mantle zone and follicle centers. Small nodules of B cells will be seen in the red pulp
♦
High-grade transformation is uncommon but occurs. In these cases, the morphology will essentially be that of diffuse large B-cell lymphoma, although evidence of residual low-grade lymphoma may be identified
♦
Bone marrow involvement may be subtle and have an intravascular pattern
Additional Studies
♦
Flow: SMZL is positive for B-cell markers (CD19+, CD20+) and typically does not express CD5, CD10, CD23, or annexin A1
♦
IHC: SMZL will be positive for B-cell markers (CD20, CD79a, PAX5) and negative for markers associated with other specific lymphoma types: CD5, CD10, bcl-6, and cyclin D1. Overexpression of p53 is associated with transformation to higher-grade tumor and a poor prognosis
♦
Molecular: IgH gene rearrangements are typically positive. Allelic loss of 7p21–32 is seen in up to 40%. Trisomy 3q and other cytogenetic abnormalities can also be seen. BRAF V600E mutations are not seen
Differential Diagnosis
♦
Benign: Splenic marginal zone hyperplasia
♦
Malignant: Other “small B-cell” lymphomas with splenic involvement including follicular lymphoma, small lymphocytic lymphoma, mantle cell lymphoma, and lymphoplasmacytic lymphoma (LPL)
Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma (Fig. 16.7)
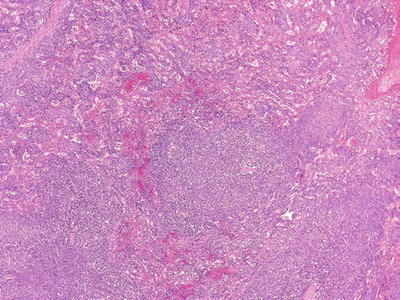
Fig. 16.7.
CLL/SLL: expansion of the white pulp with small satellite nodules. The cells are predominantly small, round lymphocytes with occasional prolymphocytes.
Definition
♦
Neoplastic proliferation of predominantly small, mature-appearing B cells that coexpress CD5 and CD19. Typically there are peripheral blood involvement (chronic lymphocytic leukemia [CLL ]) and occasionally predominant nodal involvement (small lymphocytic lymphoma [SLL])
Clinical
♦
Patients are typically >50 years old The disease is often asymptomatic but patients may have anemia, fatigue, infections, or symptomatic splenomegaly
♦
Splenic involvement can vary depending on presentation of disease. Most often splenic red pulp is prominently involved (CLL-like presentation). Rarely, the predominant involvement is in the white pulp (SLL-like presentation) with minimal red pulp involvement
Laboratory
♦
Peripheral blood and bone marrow involvement is common. The usual presentation is isolated elevated WBC composed of small, mature lymphocytes
Macroscopic
♦
Prominent white pulp involvement is associated with a grossly miliary pattern, while red pulp involvement leads to beefy appearance of the spleen. In some cases both patterns may be seen
Microscopic
♦
White pulp nodules are enlarged in size, with inconspicuous germinal centers. The predominant cell is small, round lymphocyte with scant cytoplasm and condensed chromatin. Rare larger cells with large open chromatin, prominent central nucleolus, and moderate cytoplasm (prolymphocytes, paraimmunoblasts) are present
Additional Studies
♦
Flow: CLL/SLL is positive for B-cell markers (CD19, CD20) with coexpression of CD5 and CD23. CD10 is not expressed. Some features that are typical are dim expression of CD20, no expression of FMC-7, and dim expression of surface light chains
♦
IHC: CLL/SLL has similar findings by IHC including expression of CD5, CD20, and CD23. Coexpression of CD43 and expression of bcl-2 are typical. CD10, cyclin D1, and bcl-6 are not expressed
♦
Genetics: FISH studies for genetic abnormalities are key prognostic findings in CLL/SLL. Deletions are seen at 13q14 (50%), trisomy 12 (20%), and deletions of 11q22 and 17p
♦
Molecular: Somatic hypermutation of the IgH variable region is associated with a relatively good prognosis. Cases with unmutated IgH genes appear to have a worse prognosis
Differential Diagnosis
♦
Benign: White pulp hyperplasias
♦
Malignant: Other “small B-cell” lymphomas with splenic involvement including follicular lymphoma, marginal zone, mantle cell lymphoma, and LPL
Lymphoplasmacytic Lymphoma/Waldenstrom Macroglobulinemia (Fig. 16.8)
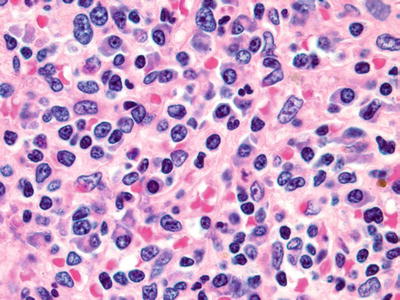
Fig. 16.8.
Lymphoplasmacytic lymphoma: red pulp infiltration by a combination of neoplastic lymphocytes, lymphoplasmacytic cells, and plasma cells.
Definition
♦
LPL is an indolent B-cell lymphoma composed of IgM-secreting cells with lymphoid and plasmacytic features. Waldenstrom macroglobulinemia is defined as LPL with bone marrow involvement and IgM monoclonal gammopathy
Clinical
♦
Some patients have symptoms of hyperviscosity caused by large amounts of serum monoclonal IgM protein. Bone marrow and peripheral blood involvement are typical. Splenic involvement is not uncommon, but the spleen is rarely removed for diagnostic purposes
♦
LPL is considered a diagnosis of exclusion; other lymphomas including marginal zone lymphoma, follicular lymphoma, and CLL/SLL can have demonstrable and sometimes abundant plasma cell differentiation. True plasma cell neoplasms must also be excluded to make a diagnosis of LPL
Macroscopic
♦
Variable and dependent on type of microscopic involvement; the patterns may be diffuse or focal with a miliary or red pulp pattern
Microscopic
♦
Most common infiltration of both white and red pulp by a mixture of small lymphocytes, lymphoplasmacytic cells, and plasma cells. Intranuclear pseudoinclusions (Dutcher bodies) and cytoplasmic immunoglobulin inclusions (Russell bodies) are not uncommon within plasma cells. Rarely, LPL may have involvement of white pulp without prominent red pulp involvement
Additional Studies
♦
Flow: LPL is positive for B-cell markers (CD19, CD20) with surface light chain restriction of the lymphocytes. FMC-7 is often positive. Typically CD5, CD10, and CD23 are negative. Plasmacytic cells will express plasma cell markers (CD38, CD138) and will have cytoplasmic light chain restriction
♦
IHC: LPL expresses B-cell markers (CD20, PAX5, CD79a). The lymphoplasmacytic cells are variably positive of plasma cell markers (CD138), while the plasma cells are positive for CD138. Cyclin D1, CD5, and CD10 are negative
♦
Molecular: IgH gene rearrangements are typically present. MYD88 mutations are seen in >90% of cases of LPL/WM, and a positive result would support this diagnosis
Differential Diagnosis
♦
Differentiation from any low-grade lymphoma with plasma cell differentiation, particularly splenic marginal zone lymphoma, can be difficult
Follicular Lymphoma (Fig. 16.9)
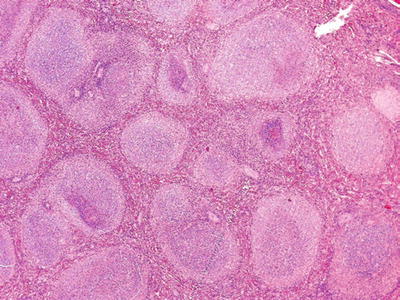
Fig. 16.9.
Follicular lymphoma: expansion and increase of white pulp/nodules with homogeneous, neoplastic lymphocytes that retain a follicular architecture.
Definition
♦
A lymphoma derived from neoplastic follicle center cells. Splenic involvement is frequent
Clinical
♦
Low-grade follicular lymphomas (FL) (grade 1 or 2) are typically indolent diseases with relentless growth. Grade 3 FL has a more aggressive clinical course and is more similar in behavior to diffuse large B-cell lymphoma. FL often presents as stage IV disease
Macroscopic
♦
Typical white pulp involvement with a miliary pattern is seen. Larger nodules (possibly with necrosis or hemorrhage) may represent sites of higher-grade transformation
Microscopic
♦
Increase in the size and density of white pulp nodules is the most prominent low power manifestation. Relatively uniform nodules are present and they are composed of predominantly small, cleaved lymphocytes (grade 1), large numbers of large lymphocytes with centroblastic features (grade 3), or only occasional large cells combined with small, cleaved cells (grade 2)
Additional Studies
♦
Flow: FL is positive for B-cell markers (CD19, CD20) with expression CD10 and restricted surface light chains
♦
IHC: Neoplastic lymphocytes express CD20, CD10, bcl-2 (75%), and bcl-6 and are negative for CD5, CD23, and cyclin D1
♦
Molecular: Rearrangement of bcl-2 gene is seen in most cases
♦
t(14;18) can be detected by FISH studies and supports a diagnosis of FL
Differential Diagnosis
♦
Benign: Follicular hyperplasia
♦
Malignant : Other “small B-cell” lymphomas with splenic involvement including small lymphocytic lymphoma, mantle cell lymphoma, and LPL
Mantle Cell Lymphoma (Fig. 16.10)
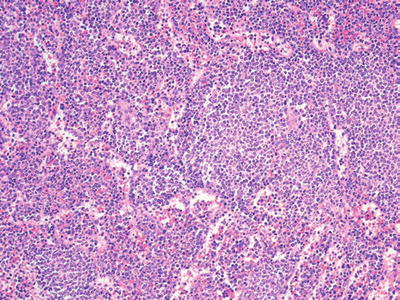
Fig. 16.10.
Mantle cell lymphoma: expansion of white pulp nodules with spillover of lymphocytes into red pulp. Cytologically, the neoplastic mantle cells are small, with irregular nuclei and condensed chromatin.
Definition
♦
A neoplastic proliferation of typically small lymphocytes , which overexpress the cyclin D1 gene product due to the presence of t(11;14). Splenic involvement is common
Clinical
♦
Often presents at high clinical stage
Laboratory
♦
Peripheral blood involvement is not uncommon
Macroscopic
♦
A miliary pattern is most common with expansion of the white pulp
Microscopic
♦
Expansion of the splenic white pulp by small lymphocytes with irregular nuclear borders. Rarely, these may be confined to an expanded mantle zone
♦
More often, abnormal lymphocytes replace the normal germinal centers, making uniform nodules. It is not uncommon for there to be “spillover” into the red pulp with clusters of neoplastic mantle cells
♦
There is a variant termed blastoid mantle cell lymphoma, which encompasses two subtypes: one with blastic nuclear chromatin and the other termed “pleomorphic,” which has larger cell size and more irregular nuclei. Blastoid variant is associated with a more aggressive clinical course and similar patterns of involvement or may occasionally have more diffuse involvement of the spleen
Additional Studies
♦
Flow: B cells coexpressing CD19 and CD5, which are negative for CD23, show light chain restriction (more often lambda), bright CD20, and sIg (compared to CLL/SLL with dim CD20 and sIg) and are positive for FMC7
♦
IHC: Neoplastic lymphocytes express CD5, CD20, bcl-2, CD43, SOX11, and cyclin D1. p53 is often positive. Ki67 is increased in the blastoid variant and associates with an aggressive clinical course. Lack of SOX11 expression has been suggested as a marker of more indolent disease. There is typically no expression of CD10, CD23, or bcl-6
♦
Karyotype: t(11;14)
♦
Molecular: Translocation of the bcl-1 gene and the Ig heavy chain gene; t(11;14). This leads to overexpression of cyclin D1, a cell cycle regulator
Differential Diagnosis
♦
Benign: Mantle zone hyperplasia (rare)
♦
Malignant: Other “small B-cell” lymphomas with splenic involvement including follicular lymphoma, small lymphocytic lymphoma, marginal zone lymphoma, and LPL
Nodal Marginal Zone Lymphoma
Definition
♦
A B-cell lymphoma arising from cells with characteristics of marginal zone cells, but not those primary to the spleen
Differential Diagnosis
♦
Splenic involvement by nodal or extranodal marginal zone lymphomas is quite rare compared to “primary” splenic marginal zone lymphoma
♦
Although there are some subtle immunophenotypic differences, most cases with splenic involvement are of the splenic type
Burkitt Leukemia/Lymphoma
Definition
♦
An aggressive, high-grade malignancy of germinal center-derived B cells
Clinical
♦
There are three clinical settings: (1) endemic type, (2) sporadic type, and (3) immunodeficiency related. Typically occurs in young patients (<20 years) except in immunodeficiency-related group
♦
Splenic involvement is not common and is present in high-stage disease
Macroscopic
♦
Miliary pattern; may have larger expansive nodules or diffuse disease, due to rapidity of cell growth
Microscopic
♦
Intermediate-sized lymphocytes expanding and replacing white pulp structures. Nuclei are generally round with small amounts of deeply staining cytoplasm. Mitotic figures and apoptotic bodies are common. Tingible-body macrophages may be present, imparting classic “starry sky” appearance. Subtle cytoplasmic vacuoles may be seen in cytologic preparations
Additional Studies
♦
Flow: Neoplastic lymphocytes express CD19, CD10, and CD20 with surface light chain restriction. TdT and CD5 are not expressed
♦
IHC: Abnormal lymphocytes are positive for CD20 and CD10, with a proliferation rate by Ki67 near 100%. There is no expression of CD5, cyclin D1, bcl-2, or TdT
♦
Karyotype: t(8;14) most common, t(2;8) or t(8;22) less common
♦
Molecular: Translocation of C-MYC gene on chromosome 8q24
Differential Diagnosis
♦
Blastic- or blastoid-appearing neoplasms including acute lymphoblastic leukemia, blastoid mantle cell, blastoid NK leukemia/lymphoma, and hepatosplenic T-cell lymphoma
Lymphoid Neoplasms Presenting Predominantly in Red Pulp
Hairy Cell Leukemia (Fig. 16.11)
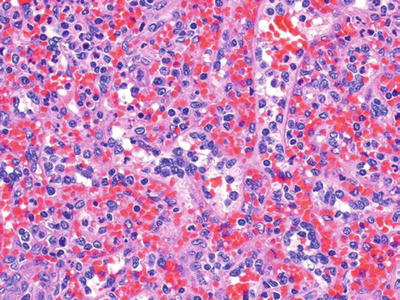
Fig. 16.11.
Hairy cell leukemia: red pulp infiltration by lymphocytes with moderate amounts of cytoplasm and relatively open chromatin.
Definition
♦
An uncommon B-cell neoplasm that prominently involves the spleen, bone marrow, and peripheral blood
Clinical
♦
Usually indolent clinical behavior
♦
Presents with symptoms related to marrow replacement (anemia) or symptoms associated with splenomegaly
Laboratory
♦
Increased circulating lymphocytes with characteristic “hairy” cytoplasmic projections
♦
These cells are positive for tartrate-resistant acid phosphatase (TRAP), which is not seen in normal lymphocytes (but may be expressed on other, low-grade B-cell lymphomas)
♦
Monocytopenia is common
Macroscopic
♦
Massive splenomegaly is common and may be presenting sign
♦
The spleen typically has a beefy appearance from expansion of the red pulp
Microscopic
♦
The red pulp is expanded by a population of abnormal cells
♦
These cells have round, centrally placed nuclei with moderate amounts of clear-to-pale cytoplasm
♦
Blood lakes (small pockets of blood in spaces surrounded by elongated hairy cells) are seen in red pulp in most cases
Additional Studies
♦
Flow: Abnormal B cells are positive for CD20, CD11c, CD22, CD25, and CD103 with light chain restriction. Annexin A1 and CD123 are also expressed. Rare examples will express CD10
♦
IHC: The lymphoid cells will express CD20, DBA.44, and TRAP. There may be some weak expression of cyclin D1. IHC staining for BRAF V600E is positive
♦
Molecular: IgH rearrangements present. BRAF V600E has been identified in almost all cases of hairy cell leukemia and the presence of this mutation supports a diagnosis
Splenic B-Cell Lymphoma, Unclassifiable
Definition
♦
A newly described entity which is a diagnosis of exclusion and includes two diagnoses, splenic diffuse red pulp small B-cell lymphoma and hairy cell leukemia variant (HC-V). This category applies to poorly defined B-cell lymphomas which do not meet the criteria of other better defined entities
Diffuse Red Pulp Small B Cell
♦
Characteristically has a leukemic presentation with massive splenomegaly. There are villous lymphocytes in the peripheral blood smears
♦
The spleen shows diffuse involvement by small B cells, which involve the cords and sinuses but spare follicular structures
♦
Immunophenotypic studies express CD20 and DBA.44. They are negative for annexin A1, CD25, CD5, CD103, CD123, cyclin D1, and CD11c
♦
BRAF V600E is not identified.
Hairy Cell Leukemia Variant
♦
This diagnosis accounts for approximately 10% of HCL, with an increased prevalence in Asian populations
♦
These cases are associated with marked lymphocytosis and involvement of bone marrow and spleen. Splenomegaly and other cytopenias are seen
♦
By morphology, these cases have hybrid features of prolymphocytic leukemia (PLL) (see below) and hairy cell leukemia (above). The bone marrow involvement may be subtle and predominantly sinusoidal in distribution
♦
Immunophenotype shows TRAP staining to be weak or negative. The cells will express CD20, CD103, FMC7, DBA.44, and CD11c. There is no expression of CD25, annexin A1, or CD123
♦
BRAF V600E is not identified
Prolymphocytic Leukemia (Fig. 16.12)
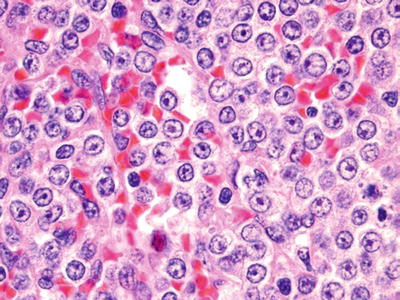
Fig. 16.12.
Prolymphocytic leukemia: in this case there is extensive red pulp infiltration by intermediate-sized lymphocytes with prominent central nucleoli.
Definition
♦
Most cases are T-cell-derived leukemias; less commonly B-cell-derived malignancies. Both are aggressive diseases and can have prominent splenic involvement
Clinical
♦
Patients present with high WBC, often over 100,000
Macroscopic
♦
Often both white and red pulp involvement
Microscopic
♦
A uniform population of intermediate to large lymphocytes They have round nuclei with prominent central nucleoli.
Additional Studies
♦
Flow: T cell: Immunophenotype is positive for CD3, CD2, and CD7 with most cases positive for CD4, coexpress CD4 and CD8 (25%), and the remainder expressing CD8
♦
B cell: Immunophenotype is positive for CD20 with bright surface immunoglobulin. There is variable expression of CD5, CD23, and FMC7
♦
IHC: Immunohistochemistry in T-cell type is positive for CD3, CD2, and CD7 with variable expression of CD4 and CD8 (as above)
♦
Immunohistochemistry in B-cell type is positive for CD20 with variable expression of CD5 and CD23 and negativity for cyclin D1 negative
♦
Karyotype: T-cell type: Abnormalities of 14q11 are common
Differential Diagnosis
♦
Need to consider blastoid variants of mantle cell lymphoma with leukemic/splenic presentation; cyclin D1 stain is necessary to evaluate the possibility of mantle cell lymphoma
Acute Lymphoblastic Leukemia
Definition
♦
A neoplasm of lymphoid precursor cells (lymphoblasts) of either B-cell origin (~90%) or T-cell origin (~10%)
Clinical
♦
Most often occur in pediatric patients. B-cell type has prominent bone marrow involvement, +/− blood involvement
♦
T-cell type often is associated with a mediastinal mass and +/− blood/bone marrow involvement
♦
Splenic involvement is common, but significant splenomegaly is rare
Laboratory
♦
Elevated WBC with blasts
Macroscopic
♦
Expansion of red pulp by neoplastic lymphoblasts in sinuses; rarely, white pulp involvement
Microscopic
♦
Intermediate-sized lymphocytes with blastic chromatin and little cytoplasm
Additional Studies
♦
Flow: Immunophenotype of B-cell type is positive for CD19 and CD10, with expression of TdT and variable positivity for CD34
♦
Immunophenotype of T-cell type is positive for CD3, CD2, CD5, and CD7. CD10 is variable. CD4 and CD8 are often both negative. TdT and CD1a are positive in some cases
♦
A berrant expression of myeloid markers may be observed in both B and T-cell types
♦
Get Clinical Tree app for offline access
Karyotype
In the T-cell type some recurrent abnormalities include involvement of 14q11.2, 7q35, and 7p14-15 (T-cell receptor genes)
In B-cell types, recurrent abnormalities are more common and better characterized
Abnormalities include:
•
t(9;22)(q34;q11.2); BCR/ABL1
•
(v;11q23); MLL rearranged
•
t(12;21)(p13;q22); ETV6/RUNX1
•
t(1;19)(q23;p13.3); PBX/E2A
•
Hypodiploid
•
Hyperdiploid > 50
•
t(5;14)(q31;q32); IL3/IGH
•
t(1;19)(q23;p13.3); E2A/PBX1

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


