Inactive Ingredients (Excipients)
Excipients are defined as ingredients added to the drug formulation for purposes other than the therapeutic or medicinally active effect. They are largely inert substances that are added to the formulation to aid in delivery of the active ingredient(s).
Well-established and well-characterized excipients are typically covered by a national or international compendium, and most pharmaceutical companies will adopt the compendial specifications and test methodology. When establishing specifications for compendial items, however, it is important to note that there may be specific attributes of the inactive ingredient that are important for the company’s product and/or manufacturing process but that are not identified as part of the compendial specifications. A good understanding of the manufacturing process, as well as knowledge of the inactive ingredient’s function in the finished dosage form, is necessary to ensure that the compendial specification captures all of the tests necessary to ensure that the inactive ingredient will perform as expected in the final drug formulation.
Intermediates
Specifications for intermediates are necessary to ensure that the material is acceptable for continued processing. Full characterization of the intermediate may not be necessary, however, and the specifications (appropriate tests and acceptance criteria) identified should be those that provide additional assurance that the final product will conform to established specifications.
Packaging Components
Packaging components are initially qualified during drug development. It is through the development process that it is shown that the selected packaging components adequately protect the product, that they do not interact with the product, and that they are constructed of materials that will not leach harmful or undesirable substances into the product during routine storage.
Once the packaging components are qualified (that is, shown to be suitable for their intended use), specifications must be established for routine quality control. Primary packaging components—those that are intended to have direct product contact—are most critical, and specifications need to be established to ensure batch-to-batch consistency of these components.
According to the United States Food and Drug Administration (FDA) guidance Container Closure Systems for Packaging Human Drugs and Biologics, quality control (QC) specifications for packaging components typically include physical characteristics (dimensional criteria, physical parameters, performance characteristics) and chemical composition (colorants, material of construction). For most drug products, a pharmaceutical company may accept a packaging component lot based on acceptable results documented on a certificate of analysis (COA) or certificate of compliance (COC) from the packaging component vendor, and the performance of an appropriate identification test. This type of reduced testing is acceptable provided that the vendor’s test data are periodically validated/revalidated (21 CFR 211.84). Additional specifications may be appropriate for components used in the packaging of injectable or inhalation products.
Finished Drug Product
Similarly to the requirements for drug substances, tests and acceptance criteria for drug products typically fall into one of two categories: universal tests (tests applicable to all drug products) and product-specific tests (tests unique to the drug product itself and/or the dosage form). Table 22.2 describes the analytical tests that are typical to chemical drug products.
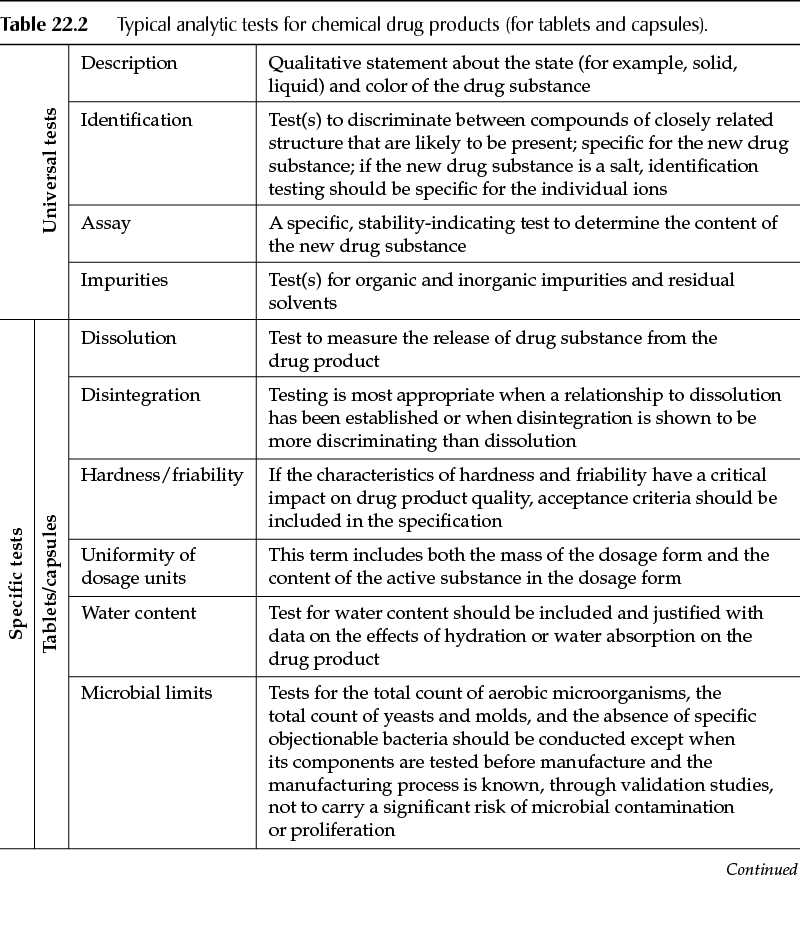
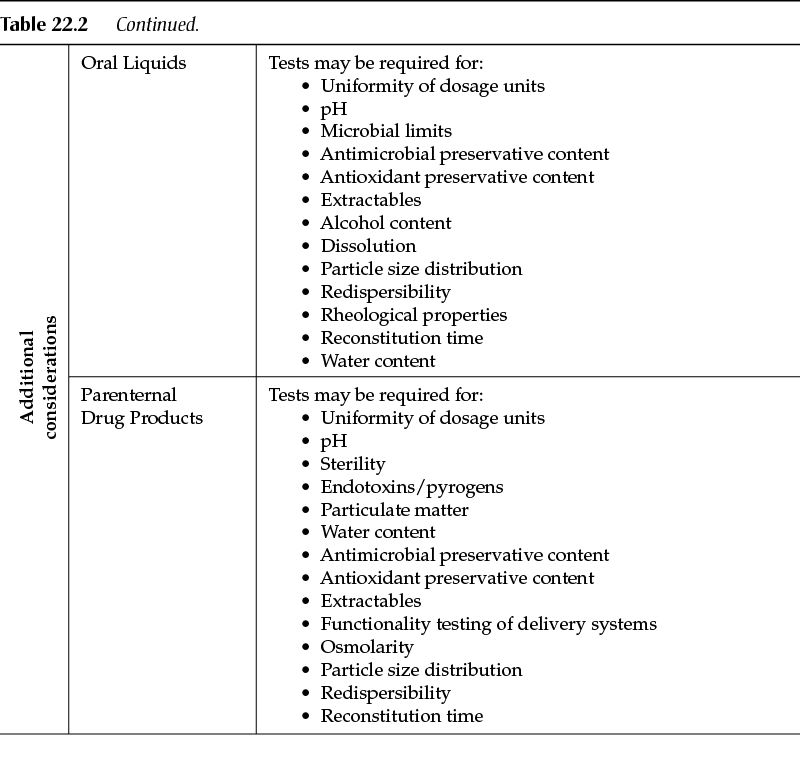
Other dosage forms (for example, oral liquids, parenteral products) will have tests specific to the type of product. Other tests may be critical, in which case the manufacturer of the drug product should have fully characterized the product during drug development and thus will be in the best position to identify the tests that are needed to ensure the quality of the finished drug product.
Finished Product Specifications
Acceptance Limits. The acceptance limits for specifications of the different quality characteristics are established by taking into account all significant elements related to the quality of the drug (medicinal) product (constancy of its characteristics), its activity (level of active constituent), and, if necessary, its safety (risk of microbial contamination, breakdown products).
Acceptance Limits of Pharmacotechnical Parameters. For most pharmacotechnical specifications, the European Pharmacopoeia, or failing this, the national pharmacopoeias of the member states, describes general test procedures with, in some cases, standards or maximal limits. In the context of these specifications, it is necessary to establish minimal and/or maximal limits, specific and adapted to the medicinal product that is the object of the marketing authorization, to guarantee the reproducibility of the finished product at manufacture.
Maximum Acceptable Deviation in Content of Active Substances. Directive 75/318/EEC states that the maximum acceptable deviation in the active substance content of the finished products shall not exceed 5% at the time of manufacture. On the basis of the stability tests, the manufacturer must propose and justify maximum acceptable tolerance limits in the active substance content of the finished product up to the end of the proposed shelf life:
• Release limits of 5% are acceptable without further justification.
• Release limits wider than 5% would need to be justified in the development pharmaceutics documentation with experimental results, which are normally based on a confidence level of 95%. The wider limits also include both the variation of the production and of the test procedure for the assay.
• Use of inadequate manufacturing procedures or inadequate test procedures (low precision) will not be accepted as a justification for wider release limits.
• To satisfy the 5% requirements, it is left to the responsibility of the manufacturer to apply an adjustment of the amount of active substance in the production of the finished product. In such a case, the overage must be clearly stated. The release limit will remain within 5% of the stated content.
• References to pharmacopoeias can not normally be accepted as a justification for wider limits as monographs do not describe a defined composition of the medicinal product, and do not pertain to release, but to recontrol by official laboratories over the whole shelf life of the product.
• Exceptionally, for certain products with a well-known degradation process and that pose no safety problems (for example, vitamins), an overage at release can be tolerated. This overage, the aim of which is to guarantee a sufficient level at the end of shelf life, must not cause an excessive level at release, and release limits must be adapted accordingly.
• At recontrol by official laboratories, the release limits of the manufacturer will be taken into consideration despite the fact that the limits used at recontrol may not be identical to the release limits of the manufacturer (due to interlaboratory variability). Where recontrol tests are carried out by another laboratory, the test procedures must be validated in their hands.
• Overfilling to guarantee the delivery of the theoretical unit dose must be justified. The acceptance limits for determination of unit content are adapted in consequence.
Acceptance Limits for Excipients
Excipients that affect the bioavailability of an active substance must be the object of a quantitative determination in each batch, unless bioavailability is guaranteed by other appropriate tests, established on a case-by-case basis as a function of development studies. In the case of preservatives, content limits of 90% to 110% at release should be acceptable without further justification, except in special cases. On expiry, the lower limit of antimicrobial preservatives may be reduced subject to the results of satisfactory preservative efficacy testing. For chemical preservatives (antioxidants), the lower limit may be considerably lower than 90% during the shelf life because of the preferential degradation of these agents.
Additional Considerations
For all incoming materials (those purchased from third-party suppliers), the specifications should clearly identify the approved suppliers for the raw material in question. Documentation specific to the approved manufacturing sites is important as many suppliers have multiple manufacturing sites, and all of them may not be approved to supply the material. Care should be taken if the material is provided by a broker or agent for the manufacturer to ensure that the materials received are coming from the approved supplier and the approved manufacturing sites.
In addition to documenting the approved supplier(s), the specifications should include a description of the approved shipping containers (these are typically the containers that the supplier has used to establish the raw material shelf life) and any special environmental conditions for maintaining the quality of the material during storage. Specifications should include the name and internal (company-specific) code for the material(s); the name should include any reference to a pharmacopoeial monograph (for example, United States Pharmacopeia [USP]).
Finally, information should be provided that is related to the sampling plan to be used to sample the raw materials and any special instructions necessary to protect the individual sampling the material, as well as any precautions to protect the material itself. Directions for sampling may be included in the specification (or reference provided to procedures that describe the sampling process).
Specification Revision
Post-approval changes (or post-launch changes in the case of non-submission products) in the laboratory are not uncommon. As the manufacturer gains experience with the drug product and the analytical methodology, changes may be necessary to ensure that the methods are robust and are capable of producing consistent and reliable analytical results. Changes made in the manufacturing process, or in the pharmaceutical product itself, may have an impact on the analytical methodology; revisions to the methodology may be necessary to accommodate these changes.
Compendial changes are not uncommon. Pharmaceutical manufacturers should have mechanisms in place to track proposed compendial changes and to evaluate the impact of the changes on applicable drug products. Pharmaceutical manufacturers need to have a robust change control process in place to capture and incorporate necessary changes.
Changes to a packaging component (for example, changes to the materials of construction or the manufacturing process used to produce the component) must be assessed and, if appropriate, reported to the applicable regulatory agency. The filing requirements vary depending on the type and impact of the change. The finished dosage form manufacturer is responsible for conducting the appropriate studies to demonstrate that the change in question does not have an adverse impact on the drug product. Specifications are updated to incorporate the change(s) once the change has been shown to be acceptable and the change has been approved by the regulatory agency, if required.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


