Markers
Tumors
Staining pattern
Comments/potential pitfalls
MDM2/
CDK4
Well-differentiated liposarcoma
Dedifferentiated liposarcoma
Intimal sarcoma
Nuclear
10% of well-dedifferentiated liposarcomas are negative by IHC and may need FISH for MDM2 amplification
MDM2 expression is seen in histiocytes
MDM2 expression alone seen in many other tumors, e.g., malignant peripheral nerve sheath tumor, solitary fibrous tumor
MUC4
Low-grade fibromyxoid sarcoma Sclerosing epithelioid fibrosarcoma
Cytoplasmic
Expressed in many different carcinomas (e.g., colon, breast) and the epithelial component of biphasic synovial sarcoma
A subset of epithelioid GISTs and myoepithelial carcinomas may show focal positivity
STAT6
Solitary fibrous tumor
Nuclear
Expressed in approximately 10% of well-differentiated and dedifferentiated liposarcomas
INI1/SMARCB1
Epithelioid sarcoma
Malignant rhabdoid tumor
50% of pediatric myoepithelial carcinomas
Loss of nuclear staining
INI1 expression is ubiquitous in nonneoplastic cells; often requires careful examination to identify negative tumor cells
TLE1
Synovial sarcoma
Nuclear
Expressed in a subset of malignant peripheral nerve sheath tumors and solitary fibrous tumors
DOG1
Gastrointestinal stromal tumor
Membranous/cytoplasmic
A subset of adenocarcinomas express DOG1
β-Catenin
Desmoid fibromatosis
Nuclear
Approximately 20% of desmoid fibromatoses are negative for β-catenin
CD31
Endothelial tumors, e.g., endothelial hemangioendothelioma, angiosarcoma
Membranous
Normally expressed in histiocytes
Approximately 10% of angiosarcomas are negative for CD31
ERG
Endothelial tumors, e.g., endothelial hemangioendothelioma, angiosarcoma
Nuclear
Highly sensitive marker of endothelial differentiation
Expressed in Ewing sarcoma with EWSR1–ERG rearrangements and prostatic adenocarcinoma with TMPRSS2–ERG fusions and 5–60% of epithelioid sarcoma depending on the antibody used
ALK
Inflammatory myofibroblastic tumor
Membranous, cytoplasmic
Expression can be seen in alveolar rhabdomyosarcoma, anaplastic large cell lymphoma
TFE3
Alveolar soft part sarcoma
Subset of PEComa
Nuclear
Also positive in renal cell carcinomas with Xp11 translocations
SOX10
Tumors with melanocytic or neuroectodermal differentiation
Nuclear
Expressed in a subset of triple negative breast cancers and salivary gland tumors
♦
Characteristic translocations and fusion genes of the most common sarcoma types are listed in Table 22.2
Table 22.2.
Common Translocations of Soft Tissue Tumors
Tumor | Translocation | Gene fusion | Frequency |
|---|---|---|---|
Alveolar rhabdomyosarcoma Alveolar soft part sarcoma | t(2;13)(q35;ql4) t(2;13)(q35;ql4) t(X;17)(p11;q25) | PAX3–FOXO1A PAX7–FOXO1A TFE3–ASPL | 65% 35% >95% |
Clear cell sarcoma | t(12;22)(ql3;ql2) t(2;22)(q33;q12) | EWSR1–ATF EWSR1–CREB1 | >90% 10% |
Dermatofibrosarcoma protuberans | t(17;22)(q21;q13) | COL1A1–PDGFB | >90% |
Desmoplastic small round cell tumor Epithelioid hemangioendothelioma | t(11;22)(p13;q12) t(1;3)(p36;q25) | EWSR1–WT1 WWTR1–CAMTA1 | >95% 90% |
Extraskeletal myxoid chondrosarcoma | t(9;22)(q22;q12) | EWSR1–NR4A3 | 75% |
Ewing sarcoma | t(11;22)(q24;q12) | EWSR1–FLI1 | 90% |
t(21;22)(q22;q12) | EWSR1–ERG | 5% | |
t(7;22)(p22;q12) | EWSR1–ETV1 | <1% | |
Infantile fibrosarcoma | t(12;15)(p13;q25) | ETV6–NTRK3 | >95% |
Inflammatory myofibroblastic tumor | Multiple, involving 2p23 | ALK fusions | >50% |
Low-grade fibromyxoid sarcoma and sclerosing epithelioid fibrosarcoma | t(7;16)(q33;p11.2) t(11;16)(p11;p11) | FUS–CREB3L2 FUS–CREB3L1 | >95% <5% |
Myxoid liposarcoma | t(12;16)(q13;p11) | FUS–DDIT3 | 95% |
t(12;22)(q13;q11) | EWSR1–DDIT3 | 5% | |
Nodular fasciitis Synovial sarcoma | t(17;22)(p13;q13) t(X;18)(p11.2;q11.2) | MYH9–USP6 SS18–SSX1 | >95% 65% |
SS18–SSX2 | 35% |
♦
Tumors of soft tissue are classified based on the line of differentiation shown by the tumor, morphologically and/or immunohistochemically; however, a subset of tumors fail to show any specific line of differentiation toward a known normal cell counterpart
♦
In addition to determining line of differentiation, knowledge of clinical context, i.e., patient age and anatomic location, is crucial in the evaluation of soft tissue tumors, as the differential diagnosis may vary greatly based on either of these demographics; for this reason, each group of tumors has been classified based on the line of differentiation and correlated with common anatomic sites of involvement (Tables 22.3, 22.4, 22.5, 22.6, 22.7, 22.8, 22.9, 22.10, 22.11, 22.12, 22.13, 22.14, 22.15, 22.16, 22.17, 22.18, and 22.19)
Table 22.3.
Classification of Fibroblastic/Myofibroblastic Tumors
Benign Keloid scar Nodular fasciitis Proliferative fasciitis Proliferative myositis Ischemic fasciitis Elastofibroma Fibroma of tendon sheath Desmoplastic fibroma Mammary-type myofibroblastoma Angiofibroblastoma Cellular angiofibroma Nuchal-type fibroma | Pediatric Fibrous hamartoma of infancy Myofibroma(tosis) Fibromatosis colli Juvenile hyaline fibromatosis Inclusion body fibromatosis Lipofibromatosis Calcifying aponeurotic fibroma Calcifying fibrous tumor Gardner fibroma |
Intermediate biologic potential Desmoid fibromatosis Inflammatory myofibroblastic tumor Solitary fibrous tumor Dermatofibrosarcoma protuberans Low-grade myofibroblastic sarcoma Infantile fibrosarcoma Palmar/plantar fibromatosis Lipofibromatosis Giant cell fibroblastoma | |
Malignant Adult fibrosarcoma Inflammatory myofibroblastic tumor Myxofibrosarcoma Sclerosing epithelioid fibrosarcoma Low-grade fibromyxoid sarcoma |
Table 22.4.
Common Anatomic Sites of Involvement of Fibroblastic/Myofibroblastic Tumors
Anatomic site | Tumor |
|---|---|
Neck | Nuchal-type fibroma Fibromatosis colli |
Shoulder | Elastofibroma |
Trunk | Dermatofibrosarcoma protuberans |
Upper extremities | Proliferative fasciitis Proliferative myositis Myxofibrosarcoma Infantile fibrosarcoma (distal) |
Hand | Fibroma of tendon sheath Calcifying aponeurotic fibroma Palmar fibromatosis Lipofibromatosis Myxoinflammatory fibroblastic sarcoma |
Inguinal/pelvic | Mammary-type myofibroblastoma Angiomyofibroblastoma Cellular angiofibroma |
Lower extremities | Myxofibrosarcoma Infantile fibrosarcoma (distal) |
Feet | Fibroma of tendon sheath Plantar fibromatosis Lipofibromatosis |
Table 22.5.
Classification of Fibrohistiocytic Tumors
Benign Benign fibrous histiocytoma Juvenile xanthogranuloma Reticulohistiocytoma Xanthoma Localized-type tenosynovial giant cell tumor Diffuse-type tenosynovial giant cell tumor Atypical fibroxanthoma Giant cell tumor of soft tissue |
Intermediate biologic potential Plexiform fibrohistiocytic tumor Giant cell tumor of soft tissue |
Table 22.6.
Common Anatomic Sites of Involvement of Fibrohistiocytic Tumors
Anatomic site | Tumor |
|---|---|
Upper extremities | Plexiform fibrohistiocytic tumor Giant cell tumor of soft tissue |
Hand | Tenosynovial giant cell tumor, localized type |
Lower extremities | Tenosynovial giant cell tumor, localized type (knee and hip) Giant cell tumor of soft tissue |
Table 22.7.
Classification of Lipomatous Tumors
Benign Lipoma Angiolipoma Spindle cell/pleomorphic lipoma Lipoblastoma(tosis) Hibernoma Lipomatosis Angiolipoma Myolipoma Chondroid lipoma Extrarenal angiomyolipoma Extra-adrenal myelolipoma |
Intermediate biologic potential Atypical lipomatous tumor/well-differentiated liposarcoma |
Malignant Dedifferentiated liposarcoma Myxoid liposarcoma Pleomorphic liposarcoma |
Table 22.8.
Common Anatomic Sites of Involvement of Lipomatous Tumor s
Tumor | Anatomic site |
|---|---|
Angiolipoma | Extremities and trunk Spindle/pleomorphic cell lipoma |
Spindle cell/pleomorphic lipoma | Neck, upper back, and shoulders |
Lipoblastoma(tosis) | Trunk and extremities |
Myolipoma | Abdominal cavity, retroperitoneum, and inguinal region |
Hibernoma | Thigh and trunk |
Atypical lipomatous tumor/well-differentiated liposarcoma | Extremities, retroperitoneum, and spermatic cord |
Dedifferentiated liposarcoma | Retroperitoneum and spermatic cord |
Myxoid liposarcoma | Extremities, most commonly the thigh |
Table 22.9.
Classification of Smooth Muscle Tumors
Benign Leiomyoma Peritoneal leiomyomatosis |
Malignant Leiomyosarcoma |
Table 22.10.
Classification of Skeletal Muscle Tumors
Benign Cardiac rhabdomyoma Adult-type rhabdomyoma Fetal-type rhabdomyoma Genital-type rhabdomyoma Rhabdomyomatous mesenchymal hamartoma |
Malignant Embryonal rhabdomyosarcoma Alveolar rhabdomyosarcoma Pleomorphic rhabdomyosarcoma Spindle cell/sclerosing rhabdomyosarcoma |
Table 22.11.
Common Anatomic Sites of Involvement of Skeletal Muscle Tumors
Anatomic site | Tumor |
|---|---|
Head and neck | Adult-type rhabdomyoma Fetal-type rhabdomyoma Embryonal rhabdomyosarcoma Spindle cell/sclerosing rhabdomyosarcoma (adult) |
Extremities | Alveolar rhabdomyosarcoma |
Inguinal/genital area | Genital-type rhabdomyoma Embryonal rhabdomyosarcoma Spindle cell/sclerosing rhabdomyosarcoma (pediatric) |
Table 22.12.
Classification of Vascular and Perivascular Tumors
Benign Papillary endothelial hyperplasia Bacillary angiomatosis Capillary hemangioma Lobular capillary hemangioma Cavernous hemangioma Epithelioid hemangioma Intramuscular angioma Angiomatosis Spindle cell hemangioma Lymphangioma | Intermediate biologic potential Kaposiform hemangioendothelioma Retiform hemangioendothelioma Papillary intralymphatic angioendothelioma Composite hemangioendothelioma Pseudomyogenic hemangioendothelioma Kaposi sarcoma Malignant Epithelioid hemangioendothelioma Angiosarcoma |
Perivascular tumors Glomus tumor Myopericytoma Angioleiomyoma |
Table 22.13.
Common Anatomic Sites of Involvement of Vascular and Perivascular Tumors
Anatomic site | Tumor |
|---|---|
Head | Epithelioid hemangioma |
Upper extremities | Glomus tumor (distal) |
Hand | Composite hemangioendothelioma Calcifying aponeurotic fibroma |
Lower extremities | Glomus tumor (distal) Intramuscular angioma Angiomatosis Papillary intralymphatic angioendothelioma (thigh) Pseudomyogenic hemangioendothelioma |
Feet | Composite hemangioendothelioma |
Table 22.14.
Classification of Peripheral Neural Tumors
Benign Traumatic neuroma Morton neuroma Digital pacinian neuroma Mucosal neuroma Solitary circumscribed neuroma Schwannoma Melanotic schwannoma Solitary neurofibroma Diffuse neurofibroma Plexiform neurofibroma Perineurioma Dermal nerve sheath myxoma Ganglioneuroma Granular cell tumor Ectopic meningioma Nasal glial heterotopia (heterotopic glial nodules) Benign triton tumor Hybrid nerve sheath tumors |
Malignant Malignant peripheral nerve sheath tumor Epithelioid malignant peripheral nerve sheath tumor Malignant triton tumor Malignant granular cell tumor Ectomesenchymoma |
Table 22.15.
Common Anatomic Sites of Involvement of Peripheral Nerve Tumor s
Anatomic site | Tumor |
|---|---|
Head and neck | Solitary circumscribed neuroma Ectopic meningioma Nasal glial heterotopia (heterotopic glial nodules) Granular cell tumor |
Extremities | Malignant granular cell tumor Malignant peripheral nerve sheath tumor Dermal nerve sheath myxoma Perineurioma |
Inguinal/pelvic | Ectomesenchymoma |
Table 22.16.
Classification of Chondro-osseous Tumors
Benign Osteoma cutis Myositis ossificans Soft tissue chondroma |
Malignant Extraskeletal osteosarcoma Extraskeletal myxoid chondrosarcoma |
Table 22.17.
Common Anatomic Sites of Involvement of Chondro-osseous Tumors
Tumor | Anatomic site |
|---|---|
Soft tissue chondroma | Hands (predominantly fingers) and feet Spindle/pleomorphic cell lipoma |
Extraskeletal osteosarcoma Extraskeletal mesenchymal chondrosarcoma | Thigh and buttocks Proximal extremities and limb girdles |
Table 22.18.
Miscellaneous Tumors and Tumors of Uncertain Differentiation
Benign Tumoral calcinosis Amyloidoma Myxoma |
Uncertain biologic potential PEComa Ossifying fibromyxoid tumor Myoepithelial carcinoma |
Malignant Alveolar soft part sarcoma Epithelioid sarcoma Clear cell sarcoma Extraskeletal Ewing sarcoma Primitive neuroectodermal tumor Extrarenal rhabdoid tumor Desmoplastic small round cell tumor Synovial sarcoma Intimal sarcoma Undifferentiated pleomorphic sarcoma |
Table 22.19.
Common Anatomic Sites of Tumors of Uncertain Differentiation
Anatomic site | Tumor |
|---|---|
Head and neck | Epithelioid sarcoma |
Trunk | Epithelioid sarcoma |
Extremities | Myoepithelial carcinoma (lower > upper) Synovial sarcoma Epithelioid sarcoma Alveolar soft part sarcoma (thigh and buttocks) Clear cell sarcoma |
Hand | Epithelioid sarcoma |
Abdomen Retroperitoneum Inguinal/pelvic | Desmoplastic small round cell tumor PEComa PEComa PEComa Epithelioid sarcoma |
♦
This chapter reviews the key features of a select group of reactive proliferations of soft tissue, as well as benign, intermediate (locally aggressive and/or rarely metastasizing), and malignant neoplasms of soft tissue
♦
Readers are referred to the suggested reading list for more detailed information on this complex group of tumors
Fibroblastic/Myofibroblastic Tumors
Benign
Keloid Scar
Clinical
♦
Keloid scarring is a reactive fibrous proliferation that almost invariably follows local trauma or surgery. It commonly affects young people of African descent; there may be a familial tendency to developing keloid scars
♦
Any anatomic location may be affected, but the head and neck region is most commonly involved
♦
Management is difficult as up to 50% of patients have local recurrence after excision
Macroscopic
♦
Keloid scarring appears as raised firm cutaneous tissue extending beyond the boundaries of the site of initial tissue damage
Microscopic
♦
Keloid scar consists of hypocellular thick, hyalinized, eosinophilic collagen bundles, haphazardly arranged into broad bands or nodules, sometimes with associated calcifications
Differential Diagnosis
♦
Hypertrophic scar, less common than keloid scarring, is confined to the original site of tissue damage and is more cellular with a nodular growth pattern and less prone to local recurrence
Nodular Fasciitis (Fig. 22.1)
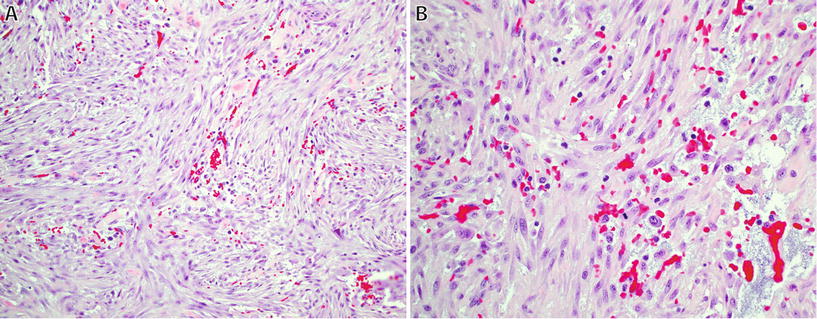
Fig. 22.1.
Nodular fasciitis: short intersecting fascicles with a “tissue culture” appearance (A), composed of plump regular spindle-shaped fibroblasts/myofibroblasts with eosinophilic cytoplasm, extravasated red blood cells, and occasional mitotic figures (B).
♦
Nodular fasciitis is a common, self-limiting, benign neoplasm composed of fibroblastic and myofibroblastic cells
♦
Histologically, nodular fasciitis is often mistaken for sarcoma
♦
Recent studies have identified a recurrent translocation t(17;22)(p13;q13) in nodular fasciitis, resulting in the formation of MYH9–USP6 fusion gene
Clinical
♦
Typically presents as a rapidly growing, often painful or tender subcutaneous nodule – most lesions are present for less than 12 weeks prior to presentation
♦
Most commonly arises in young adults but may occur at any age; no apparent gender predilection
♦
Common sites of involvement include subcutaneous tissue and fascial surfaces of the upper extremities and trunk and head and neck in children
♦
A small percentage of lesions develop in intravascular, intramuscular, and intra-articular sites; dermal involvement is rare
♦
Cranial fasciitis occurs predominantly in infants and involves soft tissues of the scalp and underlying skull bone, often resulting in bone erosion
♦
Nodular fasciitis is a self-limiting neoplasm and shows spontaneous regression if untreated; less than 2% recur locally after incomplete excision
Macroscopic
♦
Grossly, nodular fasciitis is unencapsulated and either well-circumscribed or infiltrative, usually measuring <3 cm in diameter
♦
The cut surface may have a myxoid, cystic, or fibrous appearance
Microscopic
♦
Nodular fasciitis consists of a cellular proliferation of spindled myofibroblastic and fibroblastic cells in short intersecting fascicles or whorls
♦
T he stroma is variably loose, myxoid, or collagenous with a feathery appearance and contains delicate thin-walled vessels, resembling granulation tissue
♦
Neoplastic cells have plump spindled nuclei with occasional prominent nucleoli and palely eosinophilic cytoplasm; tumor cells lack nuclear atypia and pleomorphism
♦
Mitotic forms may be numerous, but atypical forms are not seen
♦
Additional features include extravasated red blood cells, osteoclast-like multinucleated giant cells, scattered inflammatory cells, keloidal hyalinization, and, rarely, reactive new bone formation
Immunohistochemistry
♦
Tumor cells express muscle-specific actin and smooth muscle actin, consistent with myofibroblastic differentiation, and are negative for desmin, keratin, and S-100 protein
♦
CD68 expression may be seen in the osteoclast-like giant cells
Variants
♦
Fasciitis ossificans: usually periosteal in location and contains prominent reactive bone formation; it has features of both nodular fasciitis and myositis ossificans
Differential Diagnosis
♦
Sarcoma : Some sarcomas may fall into the differential diagnosis with nodular fasciitis, such as myxofibrosarcoma or myxoinflammatory fibroblastic sarcoma. In general, sarcomas rarely grow as rapidly as nodular fasciitis and, depending on the specific sarcoma type, are typically more cellular and show greater nuclear atypia and hyperchromasia. Additionally, myxofibrosarcoma also shows curvilinear vessels and pseudolipoblasts, while myxoinflammatory fibroblastic sarcoma contains Reed–Sternberg-like cells
♦
Desmoid fibromatosis: Fasciitis-like areas are often seen in intra-abdominal and breast tumors, mimicking nodular fasciitis. Fibromatosis is usually larger with a more infiltrative growth pattern, often with skeletal muscle involvement, but occasional cases can be nodular. Histologically, the fascicles of desmoid fibromatosis are longer than those in nodular fasciitis. Tumor cells are also spindled with bland nuclei, but mitoses are usually rare. 80% of desmoid fibromatoses show nuclear expression of β-catenin, which helps in the distinction from nodular fasciitis
♦
Benign fibrous histiocytoma: Usually dermal, with a mixed storiform and fascicular growth pattern with a more polymorphous population of spindled and histiocytoid tumor cells, foamy macrophages, and chronic inflammatory cells. Lateral entrapment of collagen is frequent
Proliferative Fasciitis
Clinical
♦
The most common presentation of proliferative fasciitis is a rapidly growing subcutaneous mass on the extremities (usually forearm and thigh), in patients between the ages of 40 and 70 years and without gender predilection
♦
Lesions rarely recur after excision
Macroscopic
♦
Poorly circumscribed, subcutaneous, gray-white mass, usually measuring 1–5 cm
Microscopic
♦
Histologically, this proliferation is similar to nodular fasciitis but contains numerous large ganglion-like cells, which have abundant basophilic cytoplasm and one or more round nuclei with prominent nucleoli
Differential Diagnosis
♦
Proliferative myositis: intramuscular location
♦
Nodular fasciitis: lacks ganglion-like cells; intramuscular location is unusual
Proliferative Myositis
Clinical
♦
Proliferative myositis typically presents as a solitary, rapidly growing lesion of less than 4 weeks’ duration, involving the muscles of trunk and shoulder girdle of middle-aged adults, with no gender predilection. Recurrence of the lesion is rare after excision
Macroscopic
♦
Grossly proliferative myositis is poorly demarcated and usually measures 1–6 cm in diameter. There is often scar-like induration involving muscle and overlying fascia
Microscopic
♦
The histologic appearance is similar to proliferative fasciitis, but growth between individual muscle fibers results in a classic “checkerboard-like” appearance with slight atrophy, but preservation, of muscle fibers
♦
Foci of metaplastic bone or cartilage may be present (approximately 10% of cases)
Differential Diagnosis
♦
Proliferative fasciitis: subcutaneous location
♦
Nodular fasciitis: lacks ganglion-like cells; intramuscular location is unusual
Ischemic Fasciitis (Atypical Decubital Fibroplasia)
Clinical
♦
This lesion occurs in debilitated, immobilized, or bedridden patients as the result of prolonged pressure and impaired circulation
♦
It produces a painless soft tissue mass, often mistaken for a neoplasm, in the subcutaneous areas over bony prominences such as at the shoulder, sacrum, and hip
♦
There is a slight female predilection, and the peak age is between 70 and 90 years
♦
Local recurrence after excision may occur, usually due to ongoing pressure
Macroscopic
♦
Ischemic fasciitis forms a poorly circumscribed mass in the deep subcutis, which may extend into underlying skeletal muscle
Microscopic
♦
Lobular or zonal proliferation of prominent vessels and inflamed granulation tissue with admixed plump, atypical-appearing fibroblasts
♦
Prominent myxoid stroma with cystic change, diffuse fibrinoid necrosis, and variable fibrosis (especially in older lesions)
Elastofibroma
Clinical
♦
Elastofibroma is an uncommon lesion believed to be reactive; however, reports of clonal abnormalities raise the possibility that this is a true neoplasm; clustering of cases suggests a genetic predisposition
♦
Patients are usually elderly, and there is a marked female predilection
♦
Presentation is that of a slow-growing, ill-defined mass attached to the periosteum of ribs
♦
Elastofibroma usually arises in the subscapular region and is rarely bilateral; occasional cases occur at other parts of the thoracic cavity
♦
There is no tendency for local recurrence or metastatic potential; surgery is curative
Macroscopic
♦
Elastofibroma has ill-defined lesional margins that typically measure between 5 and 10 cm
♦
The cut surface has a dense fibrous or rubbery texture with infiltration of surrounding adipose tissue
Microscopic
♦
Elastofibroma is composed of irregular bands of dense, hypocellular collagenous fibrous tissues, with characteristically thick, eosinophilic elastic fibers with a beaded or globular appearance
♦
There are often entrapped mature adipose tissue and a variably myxoid matrix
Differential Diagnosis
♦
Gardner fibroma: lacks beaded elastic fibers; tumor cells express SMA and β-catenin
♦
Scar: lacks beaded elastic fibers, with history of prior trauma/surgery
Fibroma of Tendon Sheath (Fig. 22.2)

Fig. 22.2.
Fibroma of tendon sheath : bland fibroblastic/myofibroblastic proliferation with slit-like vessels.
Clinical
♦
This benign tumor is relatively common in adults and presents as a slow-growing, painless, hard nodule attached to the tendons of hands and feet (usually on the fingers)
♦
There is a peak age between 20 and 50 years, and males are affected twice as commonly as females
♦
Up to 25% of cases recur after incomplete excision
Macroscopic
♦
Well-circumscribed, firm, and rubbery nodules attached to tendons, 1–2 cm in size
Microscopic
♦
Well-circumscribed, vaguely lobulated, and variably cellular proliferation of bland-appearing fibroblasts/myofibroblasts within dense collagenous stroma
♦
Thin, slit-like vessels are characteristic and usually more prominent at the edges of the lesion
♦
Cellularity and mitosis vary with the duration of the lesion, with earlier lesions being more cellular and mitotically active
♦
Myxoid change and chondro-osseous metaplasia may occur
♦
Tumor cells can show SMA expression, consistent with fibroblastic differentiation
Differential Diagnosis
♦
Giant cell tumor of tendon sheath: more cellular, scattered multinucleate giant cells and less hyalinized stroma
♦
Nodular fasciitis: usually not attached to tendons, more cellular, less hyalinized, and lacks slit-like vessels
♦
Fibromatosis: usually infiltrative, long sweeping fascicles of bland spindled cells; lacks slit-like vessels
Desmoplastic Fibroblastoma
♦
Desmoplastic fibroblastoma is an uncommon benign neoplasm affecting mainly middle-aged males, with a male/female ratio of approximately 3:1
♦
Patient usually presents with a painless subcutaneous nodule on the upper or lower extremities or back
♦
Tumors are typically hypocellular and composed of bland spindled- and stellate-shaped fibroblasts embedded in a collagenous and/or myxoid stroma – the lesion is usually well circumscribed but unencapsulated, although infiltration into fat is often seen
♦
Tumor cells may show expression of SMA and are negative for desmin, S100, EMA, and CD34 – scattered AE1-/AE3 positive cells may be seen
♦
Surgical excision is curative
Palisaded Myofibroblastoma of Lymph Node
♦
This is a benign myofibroblastic proliferation arising in lymph nodes, in particular inguinal nodes
♦
The lesion often resembles schwannoma due to the presence of palisading but contains distinctive amianthoid fibers or thick collagen bundles, and the lesional cells express SMA but not S-100
Mammary-Type Myofibroblastoma
Clinical
♦
Mammary-type myofibroblastoma is a benign neoplasm that shares 13q deletion, the region of retinoblastoma-1 (RB1) tumor suppressor gene, with spindle cell lipoma and cellular angiofibroma, and may show histologic overlap with these two neoplasms
♦
Mammary-type myofibroblastoma is histologically identical to myofibroblastoma of the breast but occurs in sites (mostly subcutaneous) outside the breast, most commonly along the putative “milk line,” mainly inguinal and groin areas
♦
Arises mainly in middle-aged or elderly adults and shows a male predominance
♦
Local excision is usually curative
Microscopy
♦
Mammary-type myofibroblastoma is well circumscribed and unencapsulated
♦
Composed of small ovoid–spindled cells, resembling those in spindle cell lipoma, arranged in short fascicles
♦
Collagenous stroma with prominent hyalinized collagen bundles and admixed mast cells
♦
Adipose tissue is usually present in variable amounts
Immunohistochemistry
♦
Tumor cells express desmin and CD34 and show loss of expression of retinoblastoma (Rb) protein
Differential Diagnosis
♦
Spindle cell lipoma: virtually always arises on the posterior neck/upper back as an ill-defined, dermal, or subcutaneous mass; tumor cells are negative for desmin
♦
Cellular angiofibroma: prominent round or branching vessels, often with hyalinization; tumor cells are negative for desmin
♦
Fat-forming solitary fibrous tumors: branching hemangiopericytoma-like vessels; positive for STAT6 and CD34, and negative for desmin
Angiomyofibroblastoma
Clinical
♦
Angiomyofibroblastoma arises almost exclusively in the subcutaneous tissue of the pelviperineal region, mostly in the vulva of premenopausal adult females and less commonly in scrotal or paratesticular soft tissue in males
♦
Presentation is usually as a painless, circumscribed mass
♦
Angiomyofibroblastoma is a begin neoplasm, and simple excision is curative
Microscopy
♦
Angiomyofibroblastoma is well-circumscribed with a thin fibrous pseudocapsule and prominent small thin-walled vessels throughout a loose edematous stroma
♦
This neoplasm is variably cellular, consisting of small round to spindled cells, which are concentrated around vessels
♦
Scattered binucleate and multinucleated tumor cells are usually present
♦
Epithelioid change or degenerative nuclear atypia may be seen
♦
Mature adipose tissue is present in approximately 10% of cases
Immunohistochemistry
♦
Desmin is positive in all cases; however, expression of this marker may be decreased in tumors arising in postmenopausal women – actin is usually negative
♦
Tumor cells also commonly express estrogen and progesterone receptors
Cellular Angiofibroma
Clinical
♦
Cellular angiofibroma is a rare benign neoplasm that affects mainly middle-aged adults with no gender predilection and usually arises in the superficial vulvovaginal and inguinoscrotal soft tissues
♦
Patients present with a painless mass or hydrocele (males)
♦
Simple excision is curative; local recurrence occurs in rare cases
Microscopy
♦
Cellular angiofibroma is usually well circumscribed and may have a thin fibrous pseudocapsule
♦
Variably cellular neoplasm composed of small spindle cells with oval nuclei, closely resembling the cells in spindle cell lipoma, which are distributed somewhat haphazardly or in short fascicles
♦
Characteristic prominent vasculature consisting of medium-sized thick-walled vessels with round lumina, and some variations in size are present; hyalinization of vessel walls is common
♦
Tumor stroma is variably hyalinized, myxoid, and/or edematous; scattered mast cells are usually present
♦
Intratumoral adipose tissue is present in 50% of cases
Immunohistochemistry
♦
Cellular angiofibroma is variably positive for CD34 and almost always negative for desmin
♦
Tumor cells show loss of expression of Rb
Differential Diagnosis
♦
Mammary-type myofibroblastoma: inconspicuous vessels and bundles of hyalinized collagen; positive for desmin and CD34
♦
Angiomyofibroblastoma: small thin-walled vessels and round to ovoid tumor cells; retains Rb expression
♦
Aggressive angiomyxoma: deeper location; prominent myxoid stroma; small, thin-walled vessels with bundles of myoid cells radiating from vessel walls
Nuchal-Type Fibroma
Clinical
♦
Nuchal-type fibroma is a rare, benign fibroblastic proliferation that affects mainly young and middle-aged males and may be associated with diabetes mellitus
♦
Marked preference for the posterior neck area; however, it may arise at other anatomic sites
♦
Nuchal-type fibroma often recurs but does not metastasize
Microscopy
♦
Nuchal-type fibroma is an ill-circumscribed mass involving dermal and subcutaneous tissue, composed of dense hypocellular, haphazardly arranged collagen, often with entrapment of adnexa and adipocytes and occasionally skeletal muscle
♦
Many cases show proliferation of small nerve bundles
♦
Spindled cells are usually positive for CD34
Differential Diagnosis
♦
Gardner fibroma: in many cases, nuchal-type fibroma can appear histologically indistinguishable from Gardner fibroma. Features that favor the latter diagnosis include younger patient age, relatively less cellularity, relatively thicker collagen bundles, and expression of β-catenin in the majority of cases
Calcifying Fibrous Tumor (Fig. 22.3)
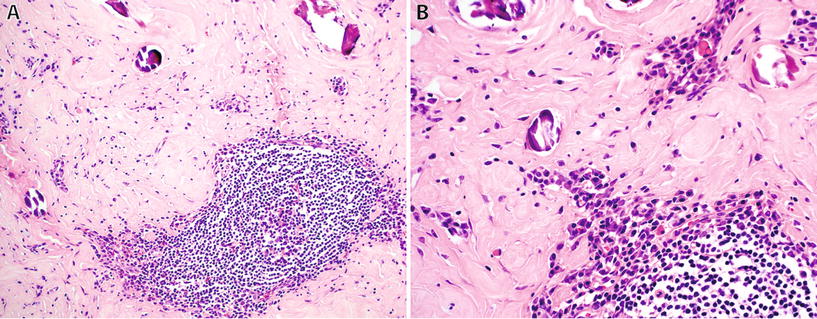
Fig. 22.3.
Calcifying fibrous tumor: well-circumscribed lesion with dense stromal sclerosis, calcifications and prominent lymphoid follicles (A), and numerous plasma cells (B).
Clinical
♦
Calcifying fibrous tumor is a benign fibrous neoplasm predominantly affecting children but also less commonly arising in adults. There is no gender predilection
♦
Most patients seek medical attention due to a painless mass in the subcutaneous and deep soft tissues of the extremities, trunk, and neck. Rarely, visceral locations and in particular peritoneal surfaces may be affected
♦
An association with inflammatory myofibroblastic tumor (IMT) has been suggested; however, this association is still controversial
Microscopy
♦
Calcifying fibrous tumor is a well-circumscribed hypocellular fibroblastic lesion characterized by a dense sclerosis, a variably prominent chronic inflammatory component with lymphoid follicles, and variable amounts of calcifications, both dystrophic and psammomatous types
♦
Examples without calcifications exist
Immunohistochemistry
♦
The spindle cells are positive for CD34 and negative for ALK
Pediatric Fibrous Tumors
Fibrous Hamartoma of Infancy
Clinical
♦
Fibrous hamartoma of infancy presents in the first 2 years of life (patients are usually younger than 1 year of age) as a solitary freely movable, poorly circumscribed soft tissue mass (3–5 cm) involving the dermis and subcutis
♦
Most common in the anterior or posterior axillary area, upper arm, and shoulder regions and show a marked male predilection
♦
Local excision is usually curative and recurrences are rare
Microscopic
♦
Fibrous hamartoma of infancy has an organoid growth pattern and is composed of three components in varying proportions: (1) myxoid foci with small round nests of undifferentiated spindle cells; (2) fascicles of myofibroblasts with wavy, tapering nuclei resembling fibromatosis; and (3) mature adipose tissue
Differential Diagnosis
♦
Lipofibromatosis: lacks the myxoid nodules of primitive cells, which are characteristic of fibrous hamartoma of infancy
Myopericytoma/Myofibroma
♦
Myofibroma and myopericytoma are benign tumors on a morphologic continuum, along with other perivascular tumors such as glomus tumor and angioleiomyoma
Clinical
♦
Myopericytoma is most commonly seen in adults, arises in the dermis or subcutis of distal extremities (>proximal extremities and head and neck), and is usually solitary but may present with multiple lesions
♦
Myofibroma occurs over a wide age range, but infants and young children (less than 2 years of age) are more commonly affected than for myopericytoma, with many cases being congenital; there is a male predominance
♦
Myofibromas may be solitary lesions or present as multicentric lesions – i.e., myofibromatosis
♦
Solitary myofibroma usually arises in the dermis or subcutis of the head and neck region, followed by trunk and extremities, and rarely in the bones of the skull
♦
Myofibromatosis may involve the soft tissue and bone, as well as visceral locations such as the gastrointestinal tract, lungs, and kidneys
♦
Although the etiology of myofibroma/myopericytoma is unknown, the occurrence of familial cases is highly suggestive of a genetic component
♦
Patients are usually cured with simple excision; recurrence is uncommon and likely represents regrowth of residual disease in multicentric or poorly circumscribed tumors. Tumors at visceral locations may have a poor outcome
Microscopic
♦
Most lesions are less than 3 cm, well-circumscribed, and unencapsulated with a lobulated or multinodular pattern. Lesions arising in deep locations and visceral sites tend to have less well-defined edges. Necrosis or cystic degenerations are common
♦
Myopericytoma is composed of ovoid to spindled cells that show concentric growth around small blood vessels and medium-sized branching hemangiopericytoma-like vessels
♦
Blood vessels near the lesion may also show a proliferation of perivascular cells
♦
Some myopericytomas show areas of conventional glomus tumor and are referred to as glomangiopericytoma
♦
Myofibroma is composed of whorls and short fascicles of bland spindled fibroblasts and myofibroblasts and plumper ovoid tumor cells, intimately associated with prominent hemangiopericytoma-like vessels
♦
The stroma is often hyalinized and may have a pseudochondroid appearance with a light blue hue
♦
Infantile myofibromatosis is often hypercellular and may mimic infantile fibrosarcoma
♦
There is often a prominent subendothelial proliferation of spindle cells with projection into vessels, which may mimic lymphovascular invasion; however, there is no association with metastasis. Mitotic activity is usually low
Immunohistochemistry
♦
Myopericytoma is positive for SMA and caldesmon
♦
Myofibroma is positive for SMA but usually negative for caldesmon
♦
Both are negative for desmin, S-100 protein, EMA, and keratins
Differential Diagnosis
♦
Glomus tumor: rounder cells with prominent cell membranes, which are highlighted by SMA and caldesmon
♦
Fibroma of tendon sheath: slit-like vessels are more prominent at the periphery of the lesion; lacks the pseudochondroid appearance of myofibroma and striking perivascular growth pattern
♦
Infantile fibrosarcoma : some cases of infantile myofibromatosis may mimic infantile fibrosarcoma; FISH or RT-PCR to detect the ETV6–NTRK3 fusion gene characteristic of the latter tumor may be needed to resolve this differential
Fibromatosis Colli
♦
Fibromatosis colli is a fusiform, scar-like fibrous replacement of the distal sternocleidomastoid muscle that is seen in neonates usually by 2–4 weeks of age, resulting in cervicofacial asymmetry (torticollis)
♦
It is often associated with abnormal positioning in utero and traumatic delivery, as well as with other musculoskeletal abnormalities
♦
Microscopically, fibromatosis colli is characterized by a variably cellular fibroblastic/myofibroblastic proliferation with a myxoid to collagenous stroma
♦
Entrapment of skeletal muscle, evidenced by the presence of scattered multinucleated muscle fibers, is invariably present
♦
Most lesions will resolve with nonsurgical approaches, such as physical therapy and stretching exercises; however, approximately 10% of cases require surgical intervention. Most patients achieve complete cure if treated before 1 year of age
Juvenile Hyaline Fibromatosis
♦
Juvenile hyaline fibromatosis is an exceedingly rare, hereditary disorder of infants and children with an autosomal recessive pattern of inheritance
♦
This disorder is characterized by cutaneous, tumorlike lesions involving the head and neck, gingiva, and periarticular soft tissue (leading to contractures) and bone, especially the skull, long bones, and phalanges
♦
Morphologically, lesions are composed of plump fibroblasts embedded in a hyaline nonfibrillary material
♦
Mutations of the gene encoding capillary morphogenesis protein 2 (CMG2) on chromosome 4q21 are implicated in the pathogenesis of this disorder
♦
Local recurrence is common; prognosis is dependent on the number, size, and location of nodules
Inclusion Body Fibromatosis (Infantile Digital Fibromatosis)
♦
This is a distinct fibrous tumor that occurs on the dorsal aspect of fingers and toes of infants as a small nodule (usually measuring <2 cm) in the dermis and subcutis
♦
Patients are usually less than 1 year of age, and there is no gender predilection
♦
Histologically, inclusion body fibromatosis consists of an ill-defined nodule of fascicles of myofibroblasts with characteristic scattered intracytoplasmic perinuclear round eosinophilic inclusions (trichrome and actin positive). There is no cytologic atypia and mitoses are scarce
♦
Surgery is the main treatment. Recurrence is seen in approximately 60% of cases and is directly correlated with the adequacy of excision
Lipofibromatosis
Clinical
♦
Lipofibromatosis affects mainly infants and young children, with a male predominance
♦
Patients present with an ill-defined, slowly growing mass involving hands and feet
♦
Lipofibromatosis is composed of two main components in varying amounts: (1) adipose tissue and (2) spindle cells arranged in long fascicles traversing the adipocytic component
♦
The major differential diagnosis is with fibrous hamartoma of infancy. Lipofibromatosis does not contain the myxoid nodules of primitive ovoid cells, which are characteristic of fibrous hamartoma of infancy
Calcifying Aponeurotic Fibroma
Clinical
♦
Calcifying aponeurotic fibroma is a rare, slowly growing fibroblastic proliferation that occurs in the distal extremities (palms and soles, wrist, and ankle) of children and adolescents (median age 9 years), with a slight male predominance
♦
Clinical presentation is with a solitary, poorly defined, painless mass arising close to or involving aponeuroses and tendons
♦
Local recurrence occurs in approximately 50% of patients
Microscopic
♦
Tumors are usually less than 3 cm and have an infiltrative growth pattern, with entrapment of adjacent normal structures
♦
Histologically, islands of central calcification are surrounded by small, round cells arranged in a linear pattern. Fascicles of fibroblasts/myofibroblasts are present between these islands
♦
Osteoclast-like giant cells and cartilaginous differentiation may be present
Gardner Fibroma
Clinical
♦
Gardner fibroma is an uncommon benign fibroblastic proliferation arising in infants, children, and teenagers, with no sex predilection. It affects mainly the paraspinal area, chest wall, flank, head and neck, and extremities
♦
Gardner fibroma is frequently associated with the development of desmoid-type fibromatosis and familial adenomatous polyposis
♦
It usually presents as an asymptomatic plaque-like thickening in superficial and/or deep soft tissue locations
Microscopy
♦
Gardner fibroma is hypocellular and composed of haphazardly arranged thick collagen bundles with cleft-like spaces
♦
Tumor cells express CD34 and β-catenin in approximately 65% of cases
Differential Diagnosis
♦
Nuchal-type fibroma: can be very difficult to distinguish histologically from Gardner fibroma. Nuchal-type fibroma occurs in older patients (adults) and compared to Gardner fibroma is relatively more cellular, the collagen bundles are less thick, and β-catenin is negative in lesional cells
Intermediate Biologic Potential
Solitary Fibrous Tumor (Fig. 22.4)
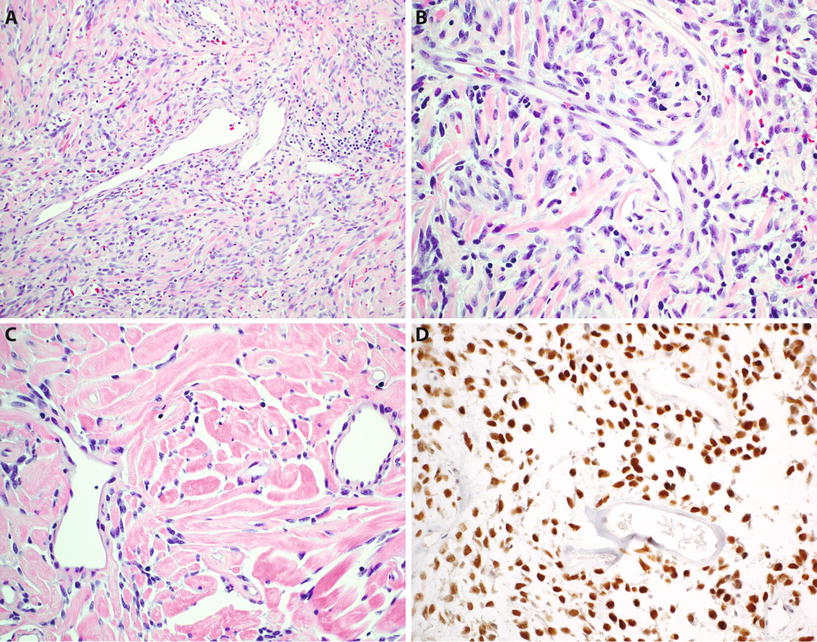
Fig. 22.4.
Solitary fibrous tumor : “patternless” spindle cell neoplasm with branching hemangiopericytoma-like vessels (A), composed of ovoid to short spindle-shaped cells with indistinct cytoplasm (B) and typically showing alternating hypercellular and hypocellular areas and hyalinized stromal collagen (C). Tumor cells show diffuse nuclear reactivity for STAT6 (D).
Clinical
♦
Solitary fibrous tumor (SFT) was originally described as a neoplasm of the pleura; however, it is now well known that SFT can arise at any anatomic location, including the visceral locations, peritoneum, retroperitoneum, mediastinum, and head and neck regions
♦
SFT arises virtually exclusively in adults, with no gender predilection, and presents as a slowly enlarging mass located in deep soft tissues
♦
Rarely associated with systemic symptoms such as hypoglycemia and finger clubbing
♦
Clinical course is variable, ranging from benign (most commonly) to malignant forms
♦
Characterized by recurrent intrachromosomal (ch12q13) translocation resulting in the formation of NAB2–STAT6 fusion gene
Microscopic
♦
SFT is usually large, well circumscribed, and vaguely lobulated
♦
A characteristic “patternless” proliferation of relatively uniform spindled tumor cells is seen, with variably prominent intervening collagen bundles
♦
There is usually marked variation in cellularity within the lesion
♦
The stroma may show marked fibrosis, especially pleural lesions, and the neoplasm has a characteristic hemangiopericytoma-like branching vascular pattern
♦
The neoplastic cells have ovoid nuclei, vesicular chromatin, inconspicuous nucleoli, and small to moderate amounts of palely eosinophilic cytoplasm
♦
Variant: lipomatous SFT is characterized by a variable adipocytic component. Most components of this variant are benign
♦
Occasionally, cases of SFT demonstrate atypical features such as diffuse hypercellularity, marked nuclear pleomorphism, or necrosis; the diagnosis of “malignant SFT” is made in the presence of four or more mitoses/10 high-power fields
Immunohistochemistry
♦
Tumor cells express STAT6 in >95% of cases, in a diffuse nuclear pattern, and show CD34 positivity in >90%
♦
Nonspecific markers CD99 and bcl2 may be positive but are nondiscriminatory from histologic mimics
♦
Negative for S-100 protein, desmin, and keratin
Fibromatoses (Figs. 22.5 and 22.6)
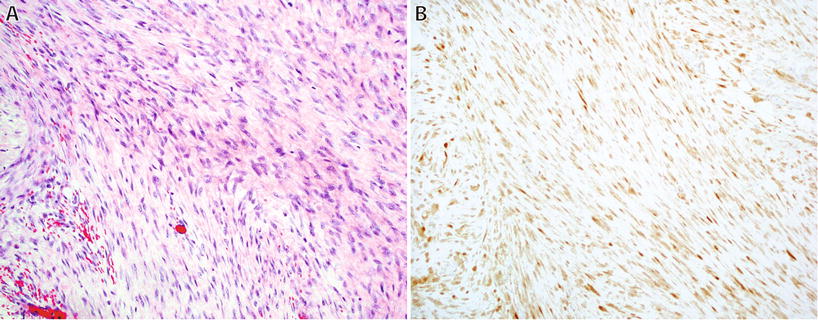
Fig. 22.5.
Desmoid fibromatosis : cellular proliferation of long sweeping fascicles of fibroblasts/myofibroblasts with fibrillary eosinophilic cytoplasm without hyperchromasia or atypia (A). Nuclear reactivity for β-catenin is present in up to 80% of cases (B).
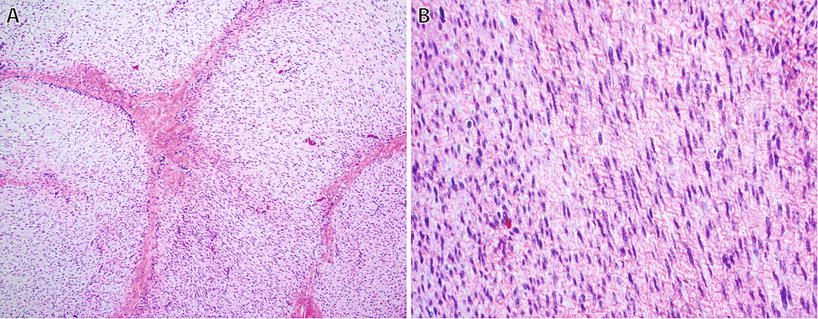
Fig. 22.6.
Plantar fibromatosis : this tumor typically has a multinodular growth pattern (A) and is hypercellular, consisting of bland spindle cells with tapering nuclei and indistinct cell borders with varying amounts of stromal collagen (B).
♦
Fibromatoses are locally aggressive neoplasms that commonly recur; however, they do not metastasize. They are subclassified into two main superficial (palmar and plantar types) and deep types
Deep (Desmoid) Fibromatoses
Clinical
♦
Desmoid fibromatosis arises in three clinical settings: sporadic, associated with familial adenomatous polyposis (FAP), and multicentric or familial types
♦
They are described on the basis of their anatomic location as follows: extra-abdominal (60%), abdominal wall (25%), or intra-abdominal (15%)
♦
The sporadic group of tumors are usually extra-abdominal, commonly involving limb girdles and proximal extremities, as well as the head and neck. Abdominal wall tumors usually occur on the anterior abdominal wall in women in the peripartum period but may also arise in a preceding scar (e.g., cesarean section). Patients with FAP tend to have intra-abdominal tumors, particularly involving the mesentery (Gardner syndrome)
♦
Peak incidence is between 15 and 40 years of age, with females more commonly affected than males
♦
In most cases, desmoid tumors are completely excised. However, in some cases, often due to anatomic location or tumor size, patients undergo conservative management, radiation therapy, or chemotherapy to attempt to shrink the tumor prior to excision.
♦
Most (85%) sporadic cases have mutations in CTNNB1, the gene that encodes β-catenin; mutations in the APC gene (chromosome 5q) are seen in patients with FAP or familial desmoids and in some sporadic cases – both mutations result in intranuclear accumulation of β-catenin
Macroscopic
♦
Grossly, desmoid tumors are usually large (>5 cm), poorly circumscribed, and infiltrative with a pale, whorled, and fibrous cut surface; intra-abdominal tumors are often well circumscribed
Microscopic
♦
Desmoid tumors are composed of long sweeping fascicles of bland fibroblasts and myofibroblasts
♦
Cellularity and mitotic activity are variable but usually low
♦
The stroma is variably collagenous with elongated, compressed blood vessels; keloidal hyalinization may also be present
♦
Some tumors may show prominent myxoid change resembling nodular fasciitis, especially tumors arising in the pelvis and mesentery
♦
Cartilaginous or osseous metaplasia occurs rarely
♦
Tumor edges are often ill defined and entrapment of, or invasion into, adjacent structures is common
Immunohistochemistry
♦
Tumor cells express actin in most cases, and nuclear β-catenin positivity is present in approximately 80% of cases
Differential Diagnosis
♦
Nodular fasciitis: looser stromal arrangement with microcystic degeneration; negative for β-catenin
♦
Palmar/plantar fibromatosis : involves palmar or plantar surfaces; multinodular growth pattern, otherwise histologically very similar to desmoid fibromatosis. SMA positive in most; 50% show nuclear expression of β-catenin. Likely genetic component to these lesions as patients often have a family history; arises in aponeuroses and results in nodules and flexion contractures of fingers. Local recurrence after surgery is common
Dermatofibrosarcoma Protuberance (Fig. 22.7)
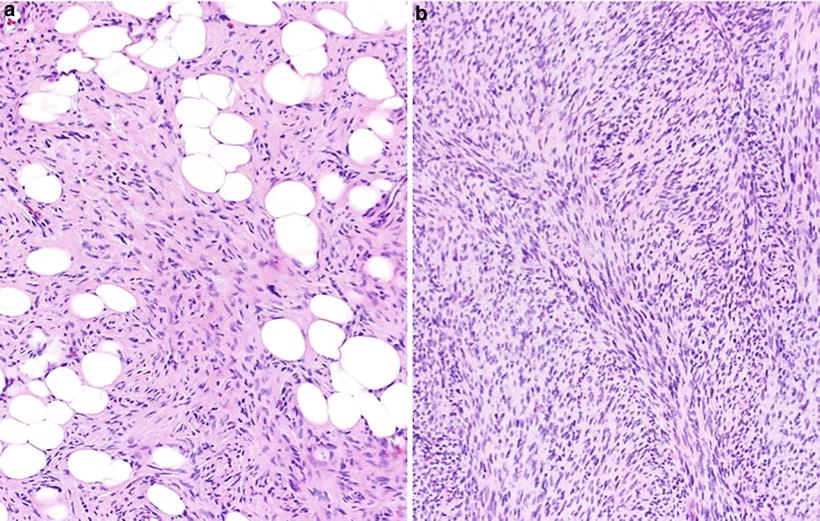
Fig. 22.7.
Dermatofibrosarcoma protuberans: storiform proliferation of monomorphic spindled cells with minimal atypia and diffuse infiltration (“honeycombing”) of subcutaneous adipose tissue (A). Fibrosarcomatous or higher-grade transformation consists of areas showing a fascicular or “herringbone” growth pattern (B).
Clinical
♦
Dermatofibrosarcoma protuberance (DFSP) involves the dermis and subcutaneous tissue of adults, between 20 and 50 years of age, without any sex or racial predilection. The most common sites of involvement are the trunk, groin, and lower extremities. They are slow-growing tumors
♦
Local recurrences are very common (30–60%), and therefore, complete local excision is required. Metastasis is uncommon and only occurs in tumors showing fibrosarcomatous transformation
Macroscopic
♦
Grossly DFSP may appear as a plaque-like lesion, nodule, or multinodular mass
♦
Cut surface is ill defined, white-gray firm mass, with an average size of 5 cm
Microscopic
♦
DFSP is a dermal-based neoplasm with diffusely infiltrative margins and extension into the subcutaneous tissue, resulting in infiltration and isolation of fat lobules, so-called “honeycombing” growth pattern
♦
The tumor is usually separated from the overlying epidermis by a narrow Grenz zone and contains entrapped adnexal structures
♦
DFSP is composed of a uniform population of fibroblast-like spindle cells arranged in a storiform growth pattern; tumor cells have plump hyperchromatic nuclei and small amounts of cytoplasm; there is minimal cytologic atypia or pleomorphism, and mitotic activity is usually low
♦
Areas of giant cell fibroblastoma may be present, usually in pediatric or young adult patients
Variants
♦
Fibrosarcomatous variant: transition to cellular fascicular growth pattern, with an increased mitotic rate and greater cytologic atypia; confers metastatic potential (15%)
♦
Bednar tumor (<5% of cases): DFSP with heavy melanin-pigmented dendritic spindle cells
♦
Myxoid variant: may mimic other myxoid neoplasms
Immunohistochemistry
♦
The neoplastic cells are positive for CD34 and negative for desmin, SMA, and S-100
Cytogenetics
♦
Supernumerary ring chromosomes derived from portions of chromosomes 17 and 22 are frequently present in adult cases of DFSP; unbalanced translocation t(17;22)(q21.3;q13.1) is present in most pediatric cases
♦
The resultant fusion gene COL1A1–PDGFB can be detected by PCR or FISH studies
Differential Diagnosis
♦
Cellular benign fibrous histiocytoma: composed of a more heterogeneous cell population (negative for CD34), often with lipid and hemosiderin deposition; shows entrapment of collagen at the edges; but lacks honeycomb pattern of infiltration of subcutaneous fat
Inflammatory Myofibroblastic Tumor
Clinical
♦
IMT is most common in childhood and adolescence; however, the age range is wide
♦
The most common sites of involvement are the lung and abdominal cavity (mesentery and omentum), but IMT has been described at nearly all anatomic sites
♦
Clinical presentation is quite variable with symptoms relating to the site of origin of the tumor – occasionally, systemic symptoms such as weight loss, fever, and hematologic abnormalities are present
♦
Local recurrence occurs in approximately 25% of patients, which is usually related to location and extent of resection; metastasis is rare, occurring in less than 5%
♦
Rearrangement of ALK gene on chromosome 2p is present in approximately 50% of tumors and of ROS1 in <5%
Microscopy
♦
IMT is a fascicular spindle cell neoplasm composed of plump fibroblasts and myofibroblasts, associated with a prominent inflammatory infiltrate rich in mainly plasma cells and lymphocytes
♦
The stroma is variably myxoid and collagenous, and the tumor is often poorly circumscribed
Immunohistochemistry
♦
The tumor cells of IMT show variable positivity for actin, desmin, and occasionally keratin
♦
ALK is expressed in approximately 50% of cases and correlates with ALK gene rearrangement, which is more commonly seen in tumors arising in childhood; a small subset of ALK-negative cases express ROS1
Differential Diagnosis
♦
Nodular fasciitis: looser storiform growth pattern, microcystic degeneration, well circumscribed, and negative for ALK
♦
Smooth muscle tumors: tumor cells have cigar-shaped nuclei and brightly eosinophilic cytoplasm and usually less inflammatory cells; SMA, desmin, and caldesmon positive and negative for ALK and ROS1
♦
Inflammatory well-differentiated/dedifferentiated liposarcoma: occurs in the retroperitoneum and may have greater cytologic atypia; tumor cells show nuclear positivity for MDM2 and CDK4
Malignant
Low-Grade Myofibroblastic Sarcoma
Clinical
♦
Low-grade myofibroblastic sarcoma occurs predominantly in young to middle-aged adults, with a slight male predilection
♦
Tumors most commonly arise in the head and neck region (tongue) but can arise at other somatic soft tissue sites, where they are usually deep seated
♦
Local recurrence is common; however, metastases are rare and tend to occur after long periods from the initial diagnosis
Microscopy
♦
Histologically, tumors are composed of hypercellular fascicles of spindled myofibroblasts with mild to moderate nuclear atypia and usually diffuse infiltration of skeletal muscle
♦
Tumors are variably positive for desmin and smooth muscle actin and are negative for S-100 protein, keratin, and caldesmon
Infantile and Adult Fibrosarcoma (Fig. 22.8)
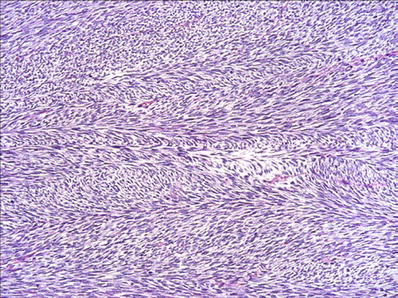
Fig. 22.8.
Infantile fibrosarcoma : “herringbone” pattern of long fascicles of monomorphic spindle cells.
Clinical
♦
Infantile fibrosarcoma is characterized by a t(12;15)(p13;q25), juxtaposing the ETV6 gene on chromosome 12 with the NTRK3 gene on chromosome 15. This is the same abnormality encountered in congenital mesoblastic nephroma and mammary analogue secretory carcinomas of the salivary gland
♦
Almost all cases of infantile fibrosarcoma arise within the first year of life and are frequently congenital; there is a slight male predominance
♦
The most commonly affected sites include the distal extremities, trunk, and head and neck regions, and presentation is with a rapidly growing soft tissue mass
♦
Recurrence rate after surgery is between 5 and 50%, and metastases are rare; the mortality rate is now less than 5%. Infantile fibrosarcoma may rarely show spontaneous regression
♦
While infantile fibrosarcoma is a clinically, histologically, and biologically distinct entity, adult fibrosarcoma is now known to actually represent/encompass several different tumor types that show a uniform fascicular growth pattern, such as malignant peripheral nerve sheath tumor (MPNST), synovial sarcoma, fibrosarcomatous component of DFSP, and low-grade myofibroblastic sarcoma
♦
A diagnosis of fibrosarcoma in adults is therefore rarely made and should be done only with caution and after excluding the above-listed tumor types – in most such cases where a line of differentiation is not identified histologically or with ancillary studies, a diagnosis of unclassified spindle cell sarcoma is more appropriate
Macroscopic
♦
Infantile fibrosarcoma is usually a large poorly defined, lobulated mass with infiltrative edges
♦
The cut surface is tan and soft with areas of myxoid and cystic degeneration, hemorrhage, and necrosis
Microscopic
♦
Infantile fibrosarcoma is a highly cellular neoplasm composed of intersecting fascicles of primitive-appearing spindle or ovoid cells forming a characteristic herringbone growth pattern
♦
There is minimal pleomorphism, with the tumor cells having a monomorphic appearance, but the mitotic rate is high
♦
Stromal collagen is variable. Necrosis, hemorrhage, and dystrophic calcifications may be present
♦
As mentioned, areas similar to fibrosarcoma may be encountered in several different tumors in adults, such as dermatofibrosarcoma protuberans and MPNST; however, areas of typical morphology are often present and help in the differential diagnosis
Immunohistochemistry
♦
There are no helpful immunohistochemical stains for infantile fibrosarcoma, and confirmation of diagnosis relies on FISH or RT-PCR to detect ETV6–NTRK3 fusion
♦
Immunohistochemistry is helpful in adult tumors with a morphologic appearance of “fibrosarcoma,” e.g., CD34 for DFSP, TLE1/EMA/AE1/3 for synovial sarcoma, and S100/GFAP/SOX10 for MPNST
Differential Diagnosis
♦
Infantile myofibromatosis: may have areas with prominent perivascular growth of spindled to ovoid tumor cells, which are SMA positive; FISH or RT-PCR to detect the ETV6–NTRK3 fusion gene of infantile fibrosarcoma may be needed to resolve this differential
♦
The tumor types that may have a fibrosarcomatous appearance in adults are listed above – in addition to IHC/molecular studies, the identification of conventional areas of a given tumor type clearly helps distinguish these tumors, e.g., DFSP
Myxoinflammatory Fibroblastic Sarcoma
♦
Myxoinflammatory fibroblastic sarcoma is a rare sarcoma associated with a translocation t(1;10)(p22:q24) that occurs in middle-aged males and females and usually involves the distal extremities, in particular hands and feet
♦
Patients present with a slowly growing, painless mass in the dermis and subcutaneous tissue
♦
Morphologically, the neoplasm shows alternating hypocellular, myxoid areas and hypercellular collagenized areas with a prominent mixed inflammatory infiltrate, and often resembles granulation tissue. Admixed atypical cells that have large nuclei and prominent eosinophilic nucleoli, resembling Reed–Sternberg (RS) cells, are key features. Areas containing macrophages filled with mucin are also present. The vascularity of myxoinflammatory fibroblastic sarcoma is inconspicuous
♦
The large RS-like cells do not show any specific immunophenotypic profile and are negative for CD30 and CD15, as well as for keratins, desmin, and S-100 protein
♦
The rate of local recurrence is as high as 70%; however, metastases are very rare (<2% of cases)
Myxofibrosarcoma (Fig. 22.9)
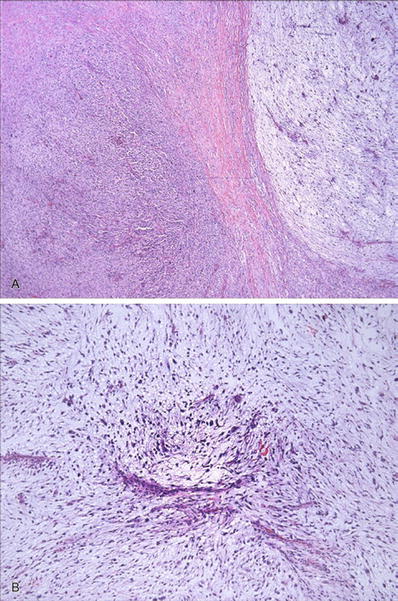
Fig. 22.9.
Myxofibrosarcoma : alternating hypercellular and hypocellular myxoid areas (A) with curvilinear blood vessels and atypical hyperchromatic cells apparent on low-power examination (B) are characteristic of this tumor.
Clinical
♦
Myxofibrosarcoma is the most common sarcoma of the extremities of older adults (>60 year of age) and shows a slight male predominance
♦
Presentation is with a subcutaneous (67% cases) or deep, slowly growing and usually painless mass
♦
The rate of local recurrence (50–60%) is directly related to the completeness of resection, while distant metastasis (20–35%) correlates with tumor depth and histologic grade
♦
Surgery is the main treatment of myxofibrosarcoma – neoadjuvant radiotherapy is often used for high-grade tumor, and adjuvant radiotherapy may be considered for positive or close margins after surgery to prevent local recurrence
♦
The most common metastatic sites are the lung and bone
Macroscopic
♦
Ill-defined, lobulated mass with poorly defined margins and variable myxoid areas
♦
Necrosis and hemorrhage are common
Microscopy
♦
Myxofibrosarcoma shows a lobulated architecture and is a variably cellular tumor: low-grade tumors are hypocellular with predominant myxoid matrix, but atypical cells can usually be readily appreciated at low-power magnification, while high-grade tumors are hypercellular with marked nuclear atypia and necrosis
♦
Curvilinear vessels are typically present and readily identified within the myxoid areas and are a helpful clue to the diagnosis
♦
Neoplastic cells tend to converge and accumulate around these vessels
♦
Nuclear pleomorphism is characteristically present as well as frequent mitoses, including atypical forms and necrosis
♦
Within the myxoid areas, pseudolipoblasts may be present and are characterized by macrophages containing mucin in the cytoplasm, without the scalloping of nuclei seen in true lipoblasts
♦
Epithelioid change may be seen and in some cases is the dominant morphology and may mimic carcinoma or melanoma; epithelioid myxofibrosarcoma is generally high grade
Immunohistochemistry
♦
IHC is generally not helpful, and its role is limited to excluding histologic mimics: tumor cells may show positivity for SMA and CD34, whereas desmin, S-100, caldesmon, and keratins are negative
♦
Myxofibrosarcoma has a complex karyotype
Differential Diagnosis
♦
Pleomorphic liposarcoma: characterized by marked pleomorphism and often mimics myxofibrosarcoma: the identification of “true” lipoblasts with atypical, hyperchromatic, indented nuclei and sharply punched-out cytoplasmic vacuoles confirms the diagnosis
♦
Pleomorphic leiomyosarcoma : composed of fascicles of spindled cells with marked pleomorphism and brightly eosinophilic cytoplasm; lacks myxoid areas and curvilinear vessels; neoplastic cells are positive for SMA, desmin, and caldesmon
♦
Pleomorphic rhabdomyosarcoma : characterized by scattered large pleomorphic tumor cells, which are positive for desmin, MYOD1 and myogenin/MYF4
Low-Grade Fibromyxoid Sarcoma (Fig. 22.10)
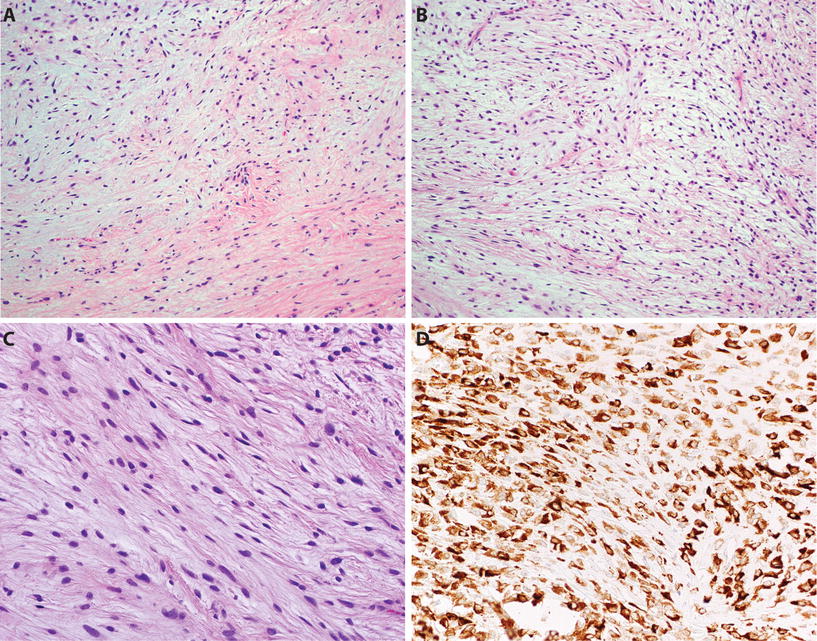
Fig. 22.10.
Low-grade fibromyxoid sarcoma : the tumor shows alternating fibrous and myxoid areas (A). Myxoid areas frequently show tumor cells arranged in short swirling fascicles (B), and tumor cells are bland with indistinct borders and bland ovoid/elongated nuclei, lacking significant atypia or pleomorphism (C). LGFMS shows diffuse cytoplasmic staining for MUC4 (D).
Clinical
♦
Low-grade fibromyxoid sarcoma (LGFMS) is a rare translocations-associated sarcoma that occurs mostly in young adults with a peak age between 30 and 50 years
♦
Most neoplasms are deeply seated, well circumscribed, and painless and occur in limbs and limb girdles
♦
The most common translocation t(7;16)(q33;p11) results in the formation of FUS–CREB3L2 fusion gene; less commonly FUS–CREB3L1 or EWSR1–CREBL1 fusion genes are present
♦
Recurrence (60%) and metastasis (6–50%) may occur many years after initial diagnosis, and patients require long clinical follow-up; metastases are commonly seen in the lungs
Microscopy
♦
The tumor is composed of swirling fascicles of bland, uniform spindled fibroblasts within abruptly alternating fibrous and myxoid areas
♦
The tumor cells are small with minimal nuclear pleomorphism and rare mitoses and may mimic benign tumors such as soft tissue perineurioma and cellular myxoma, but histologic mimics are negative for MUC4 by IHC
♦
Some cases may have large hyalinized rosettes (formerly known as hyalinizing spindle cell tumor with giant rosettes)
Immunohistochemistry
♦
MUC4 is a newly described immunohistochemical marker for LGFMS and shows diffuse cytoplasmic staining in approximately 99% of cases
♦
Tumor cells also show expression of EMA in 75% and are negative for actin, desmin, CD34, and S-100
Sclerosing Epithelioid Fibrosarcoma (Fig. 22.11)
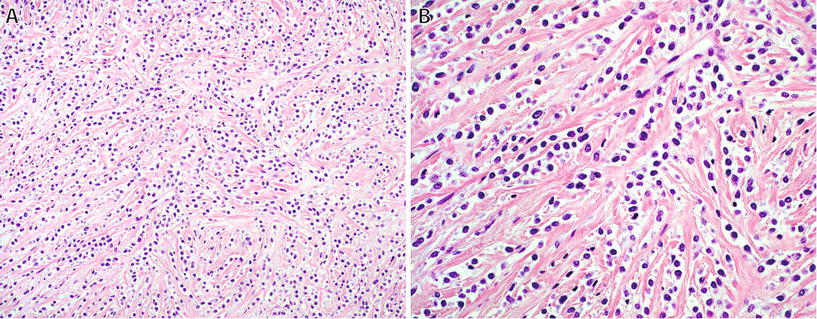
Fig. 22.11.
Sclerosing epithelioid fibrosarcoma : characteristic architecture consisting of small nests and cords (A) of polygonal epithelioid cells with pale cytoplasm in a dense sclerotic stroma (B).
♦
Sclerosing epithelioid fibrosarcoma is a rare fibroblastic sarcoma, which in some cases shares morphologic and molecular features of low-grade fibromyxoid sarcoma
♦
This high-grade tumor occurs in older adults, usually as a deep-seated mass in the extremities, and up to 50% of patients develop distant metastases
♦
The characteristic morphologic appearance is one of cords and nests of polygonal epithelioid cells with clear or palely eosinophilic cytoplasm embedded in a dense sclerotic stroma – in some cases, areas with the classic appearance of LGFMS are present
♦
This tumor may mimic carcinoma but is negative for keratins; similar to LGFMS, diffuse strong expression of MUC4 is seen in >90% of cases
♦
The most common fusion gene in sclerosing epithelioid fibrosarcoma is EWSR1–CREB3L1; a small percentage of cases have FUS–CREB3L2 fusion gene
Fibrohistiocytic Tumors
Classification of Fibrohistiocytic Tumors
Deep Benign Fibrous Histiocytoma
Clinical
♦
Benign fibrous histiocytoma may be subdivided into two main types depending on anatomic location:
Cutaneous type (most common, also known as dermatofibroma – see Chapter 18)
Deep type (<1%), arising within subcutaneous or deep soft tissue
♦
Deep fibrous histiocytoma mainly arise in adults between 20 and 40 years of age, with a slight male predominance
♦
The most common sites of involvement are the lower limb and head and neck regions; approximately 10% of cases arise in the retroperitoneum or mediastinum
♦
Local recurrence occurs in approximately 20% of cases; distant metastasis is very rare
Macroscopic
♦
Tumors are well circumscribed and pseudoencapsulated and usually measure greater than 2–3 cm; central hemorrhage or cystic change is occasionally present
Microscopic
♦
Deep fibrous histiocytoma is well circumscribed and lacks the entrapment of peripheral collagen usually seen in dermal tumors
♦
Cellular with a storiform or short fascicular growth pattern, consisting of polymorphous spindle cells with elongated or plump vesicular nuclei and indistinct cytoplasm, scattered foamy macrophages and giant cells
♦




Branching hemangiopericytoma-like vessels are common
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
