|
Typical Clinical Features |
Microscopic Features |
Ancillary Investigations |
Clear cell cutaneous fibrous histiocytoma |
Raised firm nodule on extremities, young adults |
Epidermal hyperplasia, areas of typical dermatofibroma, peripheral collagen bundles |
CK−, SMA−, desmin−, S100 protein− |
Clear cell atypical fibroxanthoma |
Sun-exposed areas, elderly patients |
Sheets of clear cells with pleomorphic nuclei, and prominent cell membranes |
CD10+ in some cases, S100 protein−, HMB45−, melan-A−, desmin−, CK− |
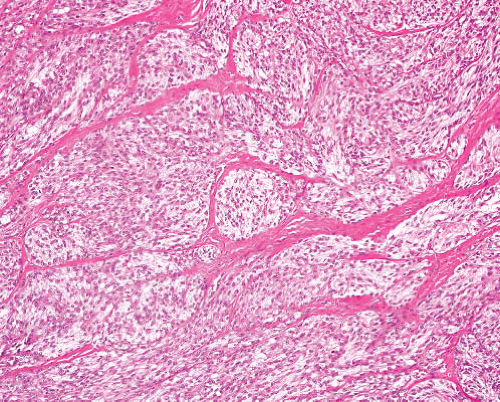
Ossifying fibromyxoid tumor |
Subcutaneous well-demarcated nodule, any age |
Partial shell of bone in many, thick fibrous capsule, cords of rounded glomus-like or occasionally spindled cells, fibromyxoid stroma
Atypical variant can have increased cellularity, mitoses, intralesional osteoid |
S100 protein+, GFAP+, desmin/SMA±, CK+ rarely, t(6;12)(p21;q24.3), EP400-PHF1 fusion |
Clear cell sarcoma (soft tissue) |
Extremities especially lower limb, young adults
In subcutis or deeper tissue, it involves tendons
Subset in gastrointestinal tract |
Nested pattern, round nuclei with central nucleolus, clear or granular cytoplasm, sometimes spindling, multinucleated cells, melanin pigment
No junctional activity |
S100 protein+, HMB45+ and melan-A+ (except in GI tract), other markers negative t(12;22)(q13;q12), EWSR1-ATF1 fusion or t(2;22)(q33;q12), EWSR1-CREB1 fusion (more often in GI cases) |
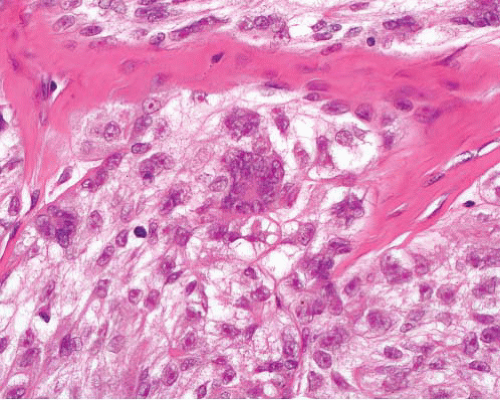
Granular cell tumor |
Subcutaneous in adults, especially in head and neck sites
Also in various organs |
Infiltrative sheets and cords of large polygonal cells, coarsely granular cytoplasm
Clear cell areas often present also |
S100 protein+, CD68+, CEA+ |
Granular cell dermatofibrosarcoma |
Like typical dermatofibrosarcoma |
Sheets of granular cells involving dermis and superficial subcutis
Areas of typical dermatofibrosarcoma usually present, with transitional forms |
CD34+, S100 protein− |
Granular cell leiomyoma |
Rare focal change in smooth muscle tumors of soft tissue or gynecologic origin |
Granular cell areas within epithelioid or spindle cell smooth muscle tumor |
SMA+, desmin+, h-caldesmon+, S100 protein− |
Paraganglioma |
In locations of paraganglia— carotid body, base of skull, mediastinum, retroperitoneum
Some are functional— hypertension, vagal symptoms |
Nests of ovoid cells with clear or granular cytoplasm and variable nuclear pleomorphism
Prominent or sinusoidal thin-walled blood vessels, sometimes fibrosis |
CD56+, NF+, SYN+, CG+. S100 protein+ in sustentacular cells |
Paraganglioma-like dermal melanocytic tumor |
Adult females, extremities, dermal nodule |
Circumscribed or infiltrative, can extend into subcutis
Nests of cells within slender fibrous septa, uniform rounded nuclei, small nucleoli, abundant clear or amphophilic cytoplasm |
S100 protein+, HMB45+, melan-A± |
Alveolar soft part sarcoma |
Childhood or adult
Single mass or multicentric
Mesenteric, retroperitoneal, other sites |
Nests of large polygonal cells with uniform vesicular nuclei and clear or finely granular cytoplasm. A solid pattern of closely packed nests is sometimes seen especially in childhood cases |
TFE3+, desmin+ der(17) t(X;17)(p11;q25), ASPSCR1-TFE3 fusion |
Rhabdomyoma |
Adult or fetal (median: 4 y) type in head and neck, or (mostly female) genital tract locations
Also cardiac rhabdomyoma (adult type) |
Sheets of large polygonal cells with abundant eosinophilic cytoplasm
Fetal rhabdomyoma has long spindle cells in myxoid stroma (immature type) or spindled and round cells with variable skeletal muscle differentiation (intermediate type) |
Desmin+, myogenin+ (in nuclei), MyoD1+ (in nuclei) |
Perivascular epithelioid cell tumor (other than organ-specific subtypes) |
Intra-abdominal, soft tissue or gynecologic locations |
Nests of ovoid or spindled cells with clear or granular cytoplasm, delicate fibrous septa
Malignant variants often epithelioid with large polygonal cells, pleomorphism, mitoses, necrosis |
SMA+, HMB45+, melan-A+, desmin±, CD117±, S100 protein+ rarely, TFE3±, cathepsin K
TFE3 gene rearrangement ± |
Chordoma |
Mainly in sacrum/coccyx, or skull base, rarely other vertebrae
Can extend into adjacent tissues as tumor mass |
Cords and nests of polygonal cells with eosinophilic cytoplasm, resembling adenocarcinoma
Larger vacuolated (physaliferous) cells
Can have areas of dedifferentiation |
S100 protein+, CK+, EMA+, brachyury+ |
Epithelioid leiomyosarcoma |
F > M
Gynecologic sites
Retroperitoneum, bowel wall or wall of vessel including inferior vena cava, renal vein |
Sheets of polygonal cells with clear or finely granular cytoplasm
Not usually pleomorphic
Typical spindle cell areas can coexist |
SMA+, desmin+, h-caldesmon+, CD117−, HMB45± |
Epithelioid gastrointestinal stromal tumor |
Related to wall of any part of alimentary tract (most commonly stomach, small intestine)
Also, in retroperitoneum, omentum |
Sheets of clear or epithelioid cells often with a focal spindle component
Organoid pattern, plasmacytoid or rhabdoid change can be seen |
CD117+, DOG1+, CD34+, h-caldesmon+. SMA variable, desmin+ rarely, S100 protein+ rarely. CK± after therapy. KIT or PDGFRA or KIT mutations |
Epithelioid pleomorphic liposarcoma |
Deep soft tissue, extremities, rarely retroperitoneum, rarely subcutis |
Sheets of clear or granular cells, pleomorphic lipoblasts
Usually focal, more typical pleomorphic liposarcoma elsewhere |
S100 protein+, AE1/3+, melan-A+, SMA+ |
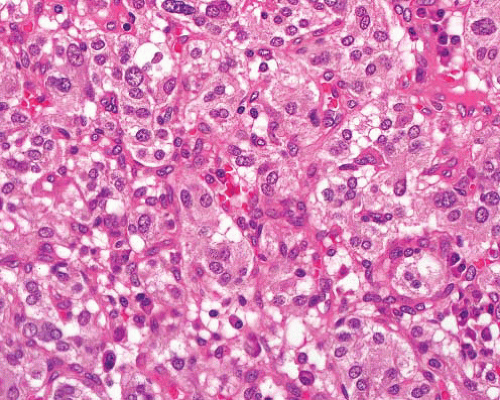
Renal cell carcinoma |
Origin in kidney, can present as metastasis |
Clear or granular cells in sheets, or adenopapillary formations
Variable nuclear grade
Can have sarcomatoid change with divergent differentiation |
CK+, EMA+, RCC+, CD117+ (oncocytoma), vimentin+ (chromophobe carcinoma) |
Adrenal cortical carcinoma |
Origin in adrenal gland
Can be functioning or nonfunctioning |
Sheets of clear or granular cells with small nuclei and occasional pleomorphism
Lacks sinusoidal vascular pattern |
Inhibin+, melan-A+, CD56+, CK− |