|
Typical Clinical Features |
Microscopic Features |
Ancillary Investigations |
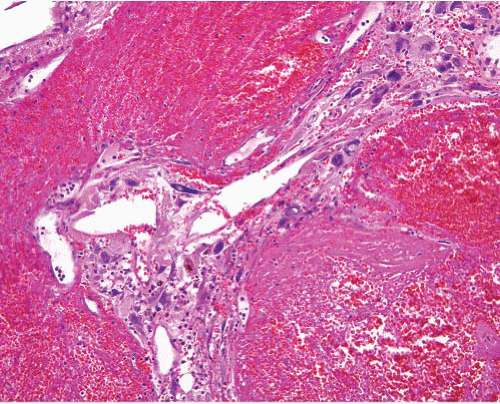
Pleomorphic hyalinizing angiectatic tumor |
Adults, solitary subcutaneous lesion, lower leg |
Circumscribed, dilated vessels with fibrinoid or hyalinized walls, atypical spindle cells, nuclear inclusions, inflammation, hemosiderin, myxoid change
No mitoses or necrosis |
CD34+ in many cases t(1;10) (p11;q24), TGFBR3-MGEA5 fusion, VGLL3, CHMP2B amplifications |
Undifferentiated pleomorphic sarcoma (malignant fibrous histiocytoma) |
Adults, deep soft tissue, proximal limbs
Intra-abdominal tumors are mostly dedifferentiated liposarcoma |
Large tumor with hemorrhage and necrosis
Pleomorphic spindle, polygonal and multinucleated tumor cells, atypical mitoses
Variable inflammation and myxoid change
Residual myxofibrosarcomatous areas in some |
Focal SMA or desmin in some cases, rare CK+ cells, CD34+ in some
MDM2+, CDK4+ in intra-abdominal examples |
Undifferentiated pleomorphic sarcoma with giant cells (giant cell malignant fibrous histiocytoma) |
Adults, deep soft tissue, proximal limbs |
As undifferentiated pleomorphic sarcoma with admixture of osteoclast-like giant cells in variable numbers, often associated with hemorrhage |
Focal SMA or desmin in some cases, rare CK+ cells. CD34+ in some
Osteoclast-like giant cells are CD68+ |
Undifferentiated pleomorphic sarcoma with inflammatory cells (inflammatory malignant fibrous histiocytoma) |
Adults, retroperitoneum and deep soft tissue of proximal limbs |
Areas of typical undifferentiated pleomorphic sarcoma, sheets of foamy cells, some atypical, heavy infiltrate predominantly of neutrophils |
Focal SMA or desmin in some cases
CD68+, in retroperitoneum many are dedifferentiated liposarcoma & MDM2+, CDK4+ |
Inflammatory well-differentiated liposarcoma |
Adults, retroperitoneum, groin |
Scattered cells with large irregular hyperchromatic nuclei in dense lymphoid infiltrate.
Areas of typical well-differentiated liposarcoma sometimes found |
MDM2+, CDK4+ |
Pleomorphic myofibrosarcoma |
Adults, deep soft tissue, proximal limbs |
Pleomorphic spindle, polygonal and multinucleated tumor cells, atypical mitoses
Variable inflammation and myxoid change |
SMA+ multifocally in subplasmalemmal “tram-track” pattern, desmin+ occasionally, h-caldesmon− |
Atypical fibroxanthoma |
Adults, head and neck, shoulder, superficial dome-shaped lesion with overlying epidermal thinning or ulceration
Dermal infiltrate is storiform, fascicular, or patternless |
Pleomorphic spindle and polygonal cells throughout, abnormal mitoses
Clear cell, spindle cell, and giant cell variants |
SMA+ focally, CD10+ |
Myxofibrosarcoma |
Subcutis, fascia, or deep soft tissue
Extremities, older adults
Recurrent |
Multinodular, spindle cells in myxoid stroma, variable nuclear pleomorphism
Can merge with high-grade pleomorphic sarcomatous areas |
CD34+ in many, SMA+ in pleomorphic areas |
Myxoinflammatory fibroblastic sarcoma |
Mostly extremities, adults
Cutaneous or subcutaneous, can involve tendons |
Multinodular ill-defined myxoid foci with vacuolated fibroblasts, intervening cellular areas with Reed-Sternberg-like and atypical mononuclear cells, eosinophils, plasma cells, hemosiderin |
CD34+ in some. t(1;10) (p11;q24), TGFBR3-MGEA5 fusion, VGLL3, CHMP2B amplifications |
Pleomorphic liposarcoma |
Adults, deep soft tissue, limbs, rarely skin or subcutis |
Undifferentiated pleomorphic sarcoma with variable numbers of pleomorphic lipoblasts, sometimes forming sheets
Rare epithelioid variant focally |
S100 protein+, CK+ (epithelioid variant), melan-A+, (epithelioid variant) |
Dedifferentiated liposarcoma |
Older adults, large retroperitoneal tumor, recurrences frequent |
Low-grade dedifferentiation: cellular fascicles with mild pleomorphism
High-grade dedifferentiation: pleomorphic undifferentiated sarcoma, or myxofibrosarcoma-like
Heterologous osteochondroid or rhabdomyosarcomatous elements
Extensive sampling might show well-differentiated liposarcomatous component, but this can be absent especially in tumors in abdomen |
MDM2+, CDK4+, P16+
Desmin, SMA, CD34 all variably focally positive
Fluorescence in situ hybridization shows MDM2 and CDK4 amplification |
Dedifferentiated chondrosarcoma |
Usually bony origin, can extend into soft tissue |
Undifferentiated pleomorphic sarcoma with contiguous differentiated chondrosarcoma of varying grade |
SMA+, S100 protein+ in chondroid area |
Pleomorphic leiomyosarcoma |
Older adults, limbs, abdomen, head and neck sites |
At least two-thirds of tumor is pleomorphic, but focally typical smooth muscle cells in fascicles are present
Pleomorphic component is undifferentiated pleomorphic sarcoma |
SMA+, desmin+, hcaldesmon+ variably in both components but less frequent in undifferentiated component |
Pleomorphic rhabdomyosarcoma |
Rare, older adults M > F, proximal limbs, retroperitoneum, pelvis |
Sheets of polygonal, round, or spindle cells with abundant eosinophilic cytoplasm, atypical mitoses, necrosis |
Desmin+, myogenin+ (nuclei), MyoD1+ (nuclei), CD56+ |
Pleomorphic mesothelioma |
Sheet-like mass involving pleura, peritoneal surface, or omentum |
Fascicles of pleomorphic spindle cells, tapered nuclei, scanty cytoplasm, mitoses, necrosis
Desmoplastic or hyalinized stroma |
CK+ focally, calretinin+ |
Undifferentiated (sarcomatoid) carcinoma |
Adults, visceral and soft tissue sites
History of primary carcinoma in some |
Pleomorphic spindle or epithelioid tumor cells in sheets, sometimes areas of epithelial morphology or overlying dysplasia |
CK+, EMA+, CD34 negative, SMA+ in some, desmin negative, h-caldesmon− |
Metastatic melanoma |
Any age, history of primary melanoma |
Pleomorphic spindle and epithelioid tumor cells, sometimes rhabdoid appearance
Melanin pigment rare |
S100 protein+ (diffuse), HMB45 and melan-A+ variably in epithelioid areas
Rare aberrant CK+ or desmin+ |
Anaplastic large cell lymphoma |
Extranodal (soft tissue) involvement is usually associated with advanced nodal disease |
Sheets of cells with prominent nucleoli, multinucleated forms |
CD30+, ALK+, CD43+, CD45+, CD3+, TIA1+, t(2;5) (p23;q35), TMP3-ALK fusion |