The estimated frequency of the various types of skeletal anomalies in children is derived from diverse sources including the experience of individual institutions, vital statistics, and registries (
44,
45). In one pediatric autopsy series that included children through 14 years of age, congenital anomalies and malformations were identified in 18% of cases; almost 20% of these were found in the skeletal system (
46). Multiple organ anomalies were found in most of these children, and major musculoskeletal anomalies were documented at autopsy in 1.3% of previable fetuses and liveborn infants who died in the perinatal period with the exclusion of chromosomal syndromes (
46). Major malformations of the limbs are found in approximately 2% of liveborn infants and minor limb abnormalities in another 5% to 7%. Overall, approximately 1:1000 neonates have some defective development of the limbs, most commonly limb reduction defects (LRDs) (
44,
46).
Among the three most common trisomy syndromes, trisomy 18 (Edwards syndrome) is characterized by overlapping fingers, radial aplasia, and other preaxial limb defects and less commonly rocker-bottom feet and equinovarus deformity, whereas trisomy 13 (Patau syndrome) has
postaxial polydactyly (
47,
48). Clinodactyly of the fifth finger with a hypoplastic middle phalanx is found in 50% to 60% of infants with trisomy 21 (
49,
50,
51). Several other musculoskeletal abnormalities are found in the setting of trisomy 21 (
52). Additional malformations of the axial skeleton in these three trisomic syndromes have been discussed elsewhere (
52,
53). Syndactyly and talipes equinovarus are the two most common limb anomalies in triploid fetuses (
53).
Limb Reduction Defects
LRDs comprise one of the most common categories of congenital skeletal anomalies and is defined by the following anatomic categories: absence or hypoplasia of a phalanx, metacarpal, or metatarsal bone as a portion of any long bone with accompanying deformity; these anomalies are represented by the specific defects of amelia, aplasia to hypoplasia of individual long bones, oligodactyly, polydactyly, and syndactyly (
Figure 28-1) (
53,
54). These developmental anomalies are seen as an isolated finding or as a component of a syndrome as one of several anomalies in other organ systems including the cardiovascular system, kidney, and intestinal tract. Approximately 70% to 75% of LRDs occur in the upper extremity, whereas 15% to 20% are detected in the lower extremity alone and both upper and lower in 10% of cases (
55,
56,
57,
58). The incidence of these defects is approximately 1:1000 to 2000 live births (
59,
60,
61). LRDs are estimated to be present in 2% of perinatal autopsies and in less than 1% of stillborns (
62). The genetic and developmental aspects of LRDs are discussed at length elsewhere (
61,
63,
64).
The morphology of LRDs includes the following anatomic categories: terminal longitudinal defects (e.g., aplasia-hypoplasia of the radius with absence of the thumb), terminal transverse defects (loss of distal limb structure with preservation of proximal structure), intercalary defects (aplasia or hypoplasia of proximal limb structure), split hand-foot defects (loss of radial ray or central ray of hand or foot), and complex defects with multiple types of LRDs. The following distribution of 271 LRDs was reported in liveborn infants from the Congenital Malformation Registry: terminal longitudinal (25%), terminal transverse (35%), intercalary (10%), split hand-foot (26%), and multiple (4%) defects (
65,
66).
The etiopathogeneses of LRDs are divisible into the following categories: dominant—recessive inheritance (15% to 20% of cases), chromosomal abnormalities (5% to 10%), known syndromes, some with a multiorgan pattern of anomalies (5% to 10%), and teratogens (3% to 5%). The latter four categories are collectively thought to account for 30% to 35% of all LRDs, and another 30% to 35% of cases are ascribed to vascular disruption. A determination as to etiopathogenesis is inconclusive for almost one-third of cases. Among those LRDs associated with congenital anomalies (12% to 33% of cases), there are patterns or associations that repeat themselves (
67). Seven specific anatomic categories of LRDs are defined in
Table 28-2. The various LRDs have several associated major congenital anomalies, some of which are better known than others (
61). Preaxial limb defects have the highest frequency (
68); they are recognized in the VATER/VACTERL association, which acronymically refers to vertebra, anorectal atresia, congenital heart, tracheoesophageal fistula, renal and distal urinary tract, and limb anomalies (
69,
70,
71,
72). These may be a consequence of perturbations in the sonic hedgehog homologue gene (7q36) and its signaling pathway since a murine knockout produces a similar pattern of anomalies as seen clinically (
73). Vertebral anomalies including vertebral fusion and butterfly vertebra as examples are present in approximately 25% of VACTERL cases, whereas limb defects are found in 10% of cases with the
preaxial absence of the radius and/or thumb and first metacarpal. A phenotype similar to VACTERL has been observed in the setting of Fanconi anemia (FA); the preaxial limb anomalies are more common in FA than in the VACTERL association (
74). Tibial aplasia-hypoplasia, another preaxial defect, is very uncommon when it is compared to the radial aplasia in the VACTERL association (
75).
Transverse limb defects with the loss of fingers and toes are associated with anorectal atresia, craniofacial anomalies, syndactyly, and genital defects. Absence of fingers and toes, cleft palate, and constriction band acrosyndactyly are anomalies associated with the
amniotic rupture sequence (ARS) or
amniotic band syndrome whose prevalence varies from 1: 1200 to 15,000 live births (
76,
77) but are commonly seen in stillbirths. Most cases of ARS are sporadic, but there is an apparent increased prevalence in type 4 Ehlers-Danlos syndrome and severe osteogenesis imperfecta (OI) (
78). Similar distal limb defects are found in association with
ventral body wall defects, which are commonly accompanied by a short umbilical cord (
79). A vascular disruption has been proposed as a possible pathogenetic factor in both ARS and ventral body wall defect (
80). Whether the tethered threads of amnion after the rupture of the amniotic sac are the entire explanation remains an unresolved issue.
Another preaxial limb defect, radial hypoplasia-aplasia, occurs in a number of syndromic settings, and it is estimated that as many as 50% to 80% of infants with absent radii have other anomalies as a component of a defined syndrome (
81,
82,
83,
84,
85) (
Table 28-3).
Lower limb deficiencies are less common than those in the upper extremities accounting for 20% to 40% of cases with or without defects in the upper extremity. Isolated deficiencies or defects of the lower extremity are uncommon, as illustrated by the fact that congenital deficiency of the tibia, or tibial hemimelia, is found in 1:1 million live births (
86,
87). Other anomalies are found in the same extremity, often other extremities and visceral organ systems in 70% to 80% of cases. Tibial hypoplasia/aplasia is found in almost 70% of cases of the VACTERL association (
70). Congenial fibular deficiency in contrast to the rarity of congenital tibial deficiency is one of the most common lower extremity deficiencies (
88).
Congenital radial-tibial deficiency is defined by an absence or hypoplasia of the preaxial structures of the extremity including the thumb, first metacarpal, radius, hallux, first metatarsal, and tibia; this anomaly is associated with nonlimb abnormalities in 70% of cases (
89). Among those are the Poland sequence and Holt-Oram syndrome (
90). Isolated femoral or fibular deficiency is equally uncommon. Somewhat more frequent is ulnar-fibular deficiency, which
is typically manifested by postaxial ray deficiency in the hands and feet with defects of the ipsilateral ulna and fibula. In most cases, ulnar-fibular deficiency is an isolated defect without anomalies elsewhere (
88).
Split hand-foot limb defect or malformation (SHFM) occurs as a sporadic (more common) or familial anomaly on the basis of a failure in the initiation and maintenance of the median apical ectodermal ridge (
91,
92). One case of SHFM is seen in 8500 to 25,000 newborns (
93). Seven chromosomal foci have been identified in isolated cases (
94). In the less common familial SHFM, both autosomal recessive and X-linked patterns of inheritance are documented (
95). Duplication in 17p13.3 (BHLHA9) and mutations FGFR1 and WNT10B have been identified (
96,
97,
98). In addition to the split hand malformation, polydactyly and syndactyly may be present. Congenital heart disease is found in almost 50% of those with an SHFM 5 mutation. Ectrodactyly, ectodermal dysplasia, and facial cleft syndrome are associated with a p63 (homologue of tumor suppressor gene, p53) mutation; p63 function is critical in the development of the limb bud and hair follicle (
99).
Patellar aplasia (absence) and hypoplasia as a lower limb deficiency are found in a number of syndromes, which are discussed at length elsewhere (
100,
101). Some of these syndromes include neurofibromatosis type 1 (NF1), campomelic dysplasia (CD) with SOX9 (17q24.3) mutations, KAT6B-related disorders, and nail-patella syndrome (NPS) with LMX1B (9q 34.1) mutation, which has a downstream effect on collagen type 4 expression in the glomerular basement development (
102). The so-called iliac horns are triangular-shaped outgrowths of the posterior ilium, which are diagnostic of NPS (see
Chapter 17).
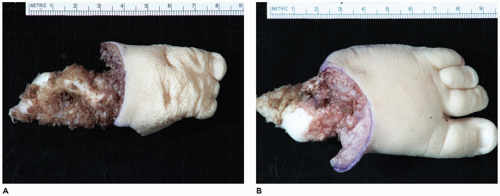
Amelia denotes incomplete or absent limb. This rare anomaly is seen in 0.15:10,000 live births and occurs with equal frequency in the upper and lower extremities (
103,
104). Amelia is associated with encephalocele, gastroschisis, omphalocele, anorectal atresia, trisomy 8, VACTERL association, and splenogonadal fusion. Severe lower limb defects are found in association with an omphalocele and diaphragmatic defect (
105). A seemingly related or similar phenotypic association is the omphalocele-exstrophy-imperforate anus-spinal defects complex with severe lower limb defects.
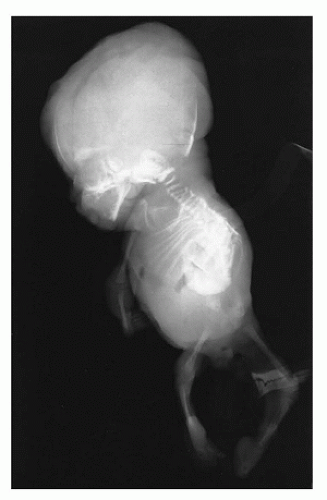
Caudal Dysgenesis
CD or
caudal regression syndrome and sirenomelia are pathogenetically related disorders of the caudal developmental field or axial mesodermal patterning (
Figure 28-2) (
106,
107,
108). Debate continues about the relationship between CD and sirenomelia (so-called mermaid syndrome) (
109,
110). Axial mesodermal dysplasia (oculo[facio]auriculovertebral spectrum and CD), CD, and sirenomelia are seen more commonly in infants of diabetic mothers to support the hypothesis of a diabetic embryopathy (
111,
112,
113,
114). However, there is no consensus whether the hyperglycemia itself is the teratogen. Limb deficiencies are another proposed manifestation of diabetic embryopathy. Dysgenesis or agenesis of the sacrum, renal agenesis, fused ectopic kidneys, ectopic ureters, müllerian duct agenesis or hypoplasia, agenesis of the bladder, cloacal exstrophy, cryptorchidism, anorectal atresia, penile-scrotal transposition, limb deficiencies, and fusion of a single dysmorphic lower limb are the range of anomalies in the genitourinary tract and lower extremities in sirenomelia (
115). The estimated frequency of CD-sirenomelia is 1:7500 births. CD has been reported in i(18q), 18p-, and trisomy 18 syndromes, VACTERL association, and heterotaxy. Retinoic acid and synthetic retinoids have been shown to cause CD experimentally. Currarino syndrome is considered a variant of caudal regression by some; hemisacrum; anorectal malformation, usually stenosis or atresia; and presacral developmental cyst are the basic phenotypic features (
116). The cyst has been interpreted as a cystic teratoma, but in some cases, it is not always clear as to the exact nature of the cyst. Hirschsprung disease and spinal dysraphia are other findings. Mutations in the homeobox gene H9 (HLXB9, MNX1 on 7q 36) have been detected in Currarino syndrome with a pattern of autosomal dominant (AD) transmission, but not in CD (
116).
Anomalies of the axial skeleton include various abnormalities in the ribs, vertebra, and sacrum. Some of these are important in their own right, whereas others are associated with more severe anomalies, such as CD including the Currarino syndrome, anorectal malformations, and
VATER/VACTERL association (
117). It has been reported that approximately 60% of those with congenital vertebral anomalies also have major or minor abnormalities in other organ systems.
Polydactyly and Syndactyly
Polydactyly is defined by the presence of six or more digits on the hand(s) or foot (feet) or both and is the obvious antithesis to the previously discussed LRDs. A defect in anterior-posterior patterning is considered the pathogenetic basis of polydactyly (
118,
119). Anatomically, similar designations to LRDs are applied to polydactyly: preaxial (lateral ray), postaxial (medial ray), and the rare central polydactyly. Polydactyly or duplication of the thumb (preaxial) is the most common example with an incidence of almost 1:100 live births. The overall prevalence is estimated to be 0.3 to 3.6:1000 live births (
119). The duplicate digit is either partially formed or a severely hypoplastic structure with minimal features to suggest a digit but rather a small polyp (
120). Histologically, the various fibrous, vascular, neural, and adipose tissues are not well organized; the peripheral nerve fibers may have traumatic neuroma-like appearance. Isolated preaxial polydactyly is more common in those of European descent, and isolated postaxial polydactyly occurs more frequently in those of African rather than European descent with incidences of 1:140-1300 live births, respectively (
119). Postaxial polydactyly in a Caucasian infant has several syndromic associations (
Table 28-4). Polydactyly can usually be observed by fetal ultrasonography at 14 to 16 weeks of gestation; one such study reported that 26 fetuses (0.15%) had polydactyly from a total of 17,760 examinations (
121).
Preaxial and postaxial polydactyly have differing genetic mechanisms by which these malformations develop. In the case of preaxial polydactyly, point mutations in sonic hedgehog are expressed along the so-called zone of polarizing activity. Postaxial polydactyly has at least three different mutated genes: 7p13, 19p 13.2, and 13q21-32; there are also frameshift mutations in GL13 (
122). The latter gene is a mediator of hedgehog signaling (
123). Two types of postaxial polydactyly have two genophenotypic expressions: type A with a well-formed digit and a normal fifth digit and type B with a hypoplastic structure resembling a small papilloma or acrochordon. When these lesions autoamputate, a traumatic neuroma is a known sequel but is less common in those hypoplastic digits, which are surgically excised. There are in excess of 300 entities in syndromic and nonsyndromic settings with polydactyly as one phenotypic feature (
118).
Syndactyly is defined by soft tissue fusion of fingers and toes with or without fusion of the small bones. Like the other anomalies in this section, syndactyly occurs as an isolated finding or as a manifestation of one of approximately 300 syndromes including acrocephalosyndactyly with its several types (Apert, Waardenburg, Pfeiffer, Summitt, and Saethre-Chotzen syndromes), Poland, Fraser, and F-syndrome (
124). Syndactyly is also a well-documented feature of the amniotic band syndrome without any specific pattern of digital or limb involvement. Polydactyly and syndactyly can also occur together with heterogeneous phenotypes.
Arthrogryposis
Arthrogryposis or congenital contracture is represented by two phenotypes: isolated or limited with single area involvement and multifocal with two or more joint contractures (
125). Multiple congenital contractures are further classified into amyoplasia, distal arthrogryposis, and related syndromes (
126,
127). The latter category includes failure in forebrain development, chromosomal abnormalities, and motor neuron disorders like spinal muscular atrophy, congenital myopathies (myosinopathies), and heritable peripheral neuropathies (
128). It has been estimated that more than 300 disorders are accompanied by multiple joint contractures and their arthrogryposis is not a specific diagnosis, but rather a phenotype (
129). The contractures are often symmetrical in both upper and lower extremities in over 50% of cases (
130) (
Figure 28-3).
Fetal akinesia-hypokinesia deformation (FAD) sequence has an estimated prevalence of 1:12,000 to 19,000 live births, and it can be recognized after the first trimester (
131,
132). These infants have the so-called Pena-Shokeir phenotype
with limb contractures (arthrogryposis), intrauterine growth restriction, an attenuated umbilical cord because of diminished fetal activity, secondary pulmonary hypoplasia, and craniofacial anomalies (
133,
134). The FAD sequence is itself a clinical phenotype with several specific genetic mutations including the Escobar syndrome (multiple pterygium syndrome) with multiple mutations involving the gamma subunit gene (CHRNG) of acetylcholine receptor (
135,
136).
Genetic Skeletal Disorders
GSD or skeletal dysplasias are the encompassing designation for the 40 groups of conditions that affect the normal development of bones and supporting tissues in terms of their shape and size and often with a reduction in normal stature (
137). The number of recognized GSDs currently comprises some 456 conditions as of the 2010 revision of the Nosology and Classification of Genetic Skeletal Disorders (
138). Among the 456 disorders, almost 70% are associated with mutations in 216 different genes. The 2010 revision was expanded to include 40 groups as defined by similar mutational and/or phenotypic characteristics (
Table 28-5). These 40 groups are arranged in clusters: (a) groups 1 to 8 by common underlying gene or pathway defect; (b) groups 9 to 17 by specific bone structure or segment involvement by imaging studies; (c) groups 18 to 20 by macroscopic criteria and clinical features; (d) groups 21 to 25 and 28 by altered bone density, mineralization, stippling, or osteolysis; (e) group 27 by lysosomal disorders with skeletal manifestations; (f) group 29 by exostosis or enchondromas (ECs); (g) group 23 or OP group; (h) group 25 or OI group; (i) group 26 by hypophosphatemic rickets; (j) group 29 by disorganized skeletal development; (k) group 30 with overgrowth including skeleton; (l) group 31 by genetic inflammatory disorders involving bones and joints; and (m) groups 32 to 40 or dysostoses with abnormalities in individual bones or groups of bones (
139).
The prevalence rate of the skeletal dysplasias is approximated at 2.4 to 34.5:10,000 stillbirths and live births, but among infants who died in the perinatal period, the frequency is higher at 9 to 10:1000 perinatal deaths (
140,
141,
142,
143,
144). Several types of skeletal dysplasias are inconsistent with survival beyond the neonatal or early infancy period and are collectively referred to as lethal chondrodysplasias (
145,
146) (
Table 28-6). The point prevalence at birth of the lethal chondrodysplasias was 15.4:100,000 births in one geographic region of Denmark (
145,
147). Whether the particular clinical observations had been obtained from prenatal diagnosis by ultrasonography or perinatal autopsies, thanatophoric dysplasia (TD) and OI type 2 are the most common lethal skeletal dysplasias, accounting for 50% to 65% of cases (
148,
149,
150) (
Table 28-7). Short-rib dysplasias (SRDs), achondrogenesis, and CD comprise the next most common lethal disorders (
126,
132). A somewhat different experience in the context of
International Skeletal Dysplasia Registry is based on referral cases with the following distribution: OI type 2 (20% of all cases), TD (11%), achondrogenesis type 2 (8%), CD (4%), and other specific disorders (36%) (
157). Approximately 4.5% of cases were unclassified. In virtually all of the lethal GSDs, there is a severe narrowing or reduction in the volume of the thoracic cavity with restricted lung growth and resulting secondary pulmonary hypoplasia (
158,
159).
Postmortem examination in GSDs. Although it may seem obvious, radiographs with anterior-posterior and lateral views should be obtained as a prerequisite to the postmortem examination on any dysmorphic infant, including one with a suspected skeletal dysplasia (
160). No conventional autopsy can hope to demonstrate the entire range of abnormalities in the skeletal system without a total body image (
161,
162). In fact, the GSDs were largely classified on the basis of their radiographic features, but molecular genetic and biochemical studies have served as the basis for classification in the majority of cases (
138).
Acquisition of tissues, mainly soft tissues rich in fibroblasts, is recommended for standard metaphase cytogenetics. Although a few hours may have lapsed since death, it is still possible to obtain cellular growth, provided that the body has been placed in a temperature-controlled environment. Samples of cartilage at the costochondral junction or joint space can be snap frozen in liquid nitrogen. The utility of standardized sections from various specific sites for optimal pathologic examination has been discussed by Yang and associates (
163).
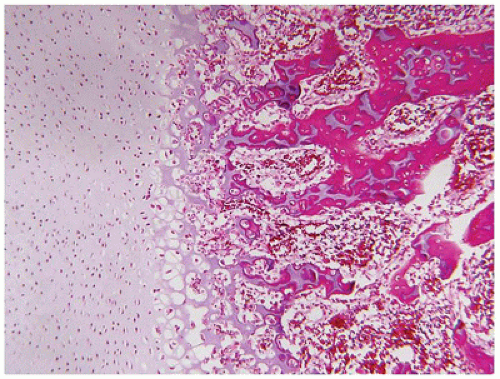
Before the internal examination is performed, a careful documentation of the various standard measurements in the perinatal autopsy and photographs from the anterior, posterior, and lateral profiles should be obtained (
164). Various sites, with particular emphasis on the regions of the growth plate, have been recommended for the sampling of membranous bone, including the ribs, vertebral bodies, proximal and distal humerus and/or femur, and cranium (
163,
165). The costochondral junctions of the fourth through sixth ribs are regarded by some as the optimal sites for identifying disturbances in the growth plate (
166,
167). Decalcified and undecalcified sections have complementary value. It is helpful to have microscopic sections available from the osteochondral junction of an age-matched infant without any known skeletal abnormalities for purposes of reference and orientation. A particularly useful review of the morphologic aspects of the growth plate has been provided by Brighton (
26). Many of the histologic abnormalities in a GSD are semiquantitative, in addition to individual cellular alterations. The cellularity of the various zones of cartilage (resting, proliferating, and hypertrophic) and their organization into columns in the hypertrophic zone and the actual chondroosseous junction or zone of provisional ossification are the specific foci of histologic interest in this group of disorders. In some but not all disorders, the morphologic abnormalities are consistent from one case to another within a specific diagnostic entity. The discussion of chondrodysplasias by Gilbert-Barness with its high-quality images, which correlate the radiographic, gross, and microscopic features, is recommended (
168).
The following discussion of GSDs is based on the various “groups” as defined in the 2010 revised classification (
138). Selected groups are considered based upon their frequency and models of morphologic and molecular pathology.
FGFR3 Chondrodysplasias (Group 1)
FGFR3 chondrodysplasia group is characterized by short limbs relative to a somewhat longer trunk. The individual entities in this group are achondroplasia (ACH), severe ACH with developmental delay and acanthosis nigricans (SADDAN), hypochondroplasia (HCP), hypochondrodysplasia-like dysplasia, and thanatophoric (TD) types 1 and 2 (
169,
170,
171). Several mutations have been identified in the FGFR3 gene (4p16.3); the germ-line mutations in FGFR3 gene inhibit chondrocyte proliferation (
172).
Achondroplasia, the most common type of chondrodysplasia, is a nonlethal disorder in the heterozygote (AD inheritance), with a birth prevalence of 1:10,000 to 30,000 live births (
173). Most cases are sporadic, with greater than 80% of cases representing a new mutation. Approximately 50% of FGFR3 chondrodysplasias are examples of ACH (
144). There is a gain of FGFR3 function, which at the growth plate has an arresting effect upon the chondrocytes with the development of rhizomelic shortening of the extremities. It has been reported that the mutation impairs endochondral bone growth by preventing SOX9 downregulation (
174). Morphologically, the growth plate is regular with periosteal overgrowth.
Hypochondroplasia is also a nonlethal disorder with AD inheritance and a prevalence of 1.5:100,000 live births. The point mutations on the FGFR3 gene (p. ASN540 Lys) in 70% to 75% of cases differ from ACH (p.Glu 380 Arg); other mutations in the FGFR3 gene have been identified in HCP (
175). The clinical and radiographic heterogeneity of HCP is substantial to serve as a challenge in the diagnosis. Compound carriers of the heterozygous mutations on the FGFR3 gene (G380R and N540K) appear to have a more morbid phenotype than those with either one or other point mutations (
176). Like ACH, the growth plate is more or less normal appearing. The latter is not surprising in that there is considerable phenotypic overlap between ACH and HCP (
176,
177).
Thanatophoric dysplasia occurs in 1:20,000 to 60,000 births and is the most common type of lethal chondrodysplasia in most series based upon prenatal ultrasonography and/or autopsy (
178,
179,
180,
181,
182,
183). Like the other FGFR3 opathies, TD has AD inheritance. Two phenotypes of TD are recognized: type I with curved femora and missense point mutations, p.Arg 248 Lys and p.Tyr 373 Lys (90% of cases), and type II with straight femora and cloverleaf skull with the exclusive mutation, p. Lys 650 Glu (100% of cases) (
176,
178). Approximately 80% to 85% of TD cases are type I and the remainder are type II. In the ISDR with mutational analysis of the FGFR3 opathies, 65% of the cases were examples of TD types I and II, ACH (25%), and HCP (9%) (
176).
Another phenotype of the FGFR3 opathies is severe achondrodysplasia with developmental delay and acanthosis nigricans (SADDAN) (
184,
185). In SADDAN, the point mutation is at codon 650 (p.Lys 650 Net) (
184). Acanthosis nigricans and epidermal nevus are other expressions of FGFR3 mutations (
186).
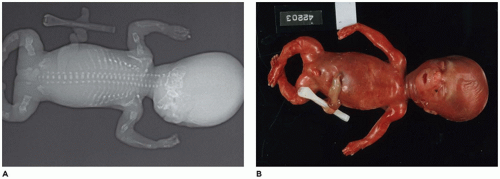
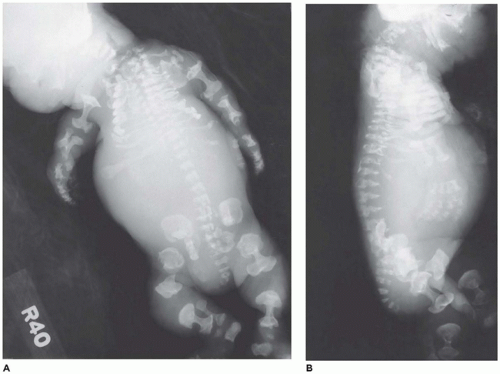
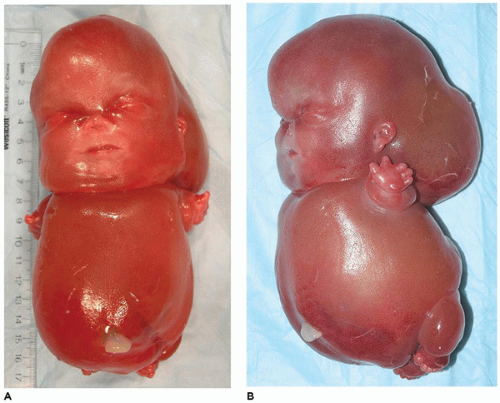
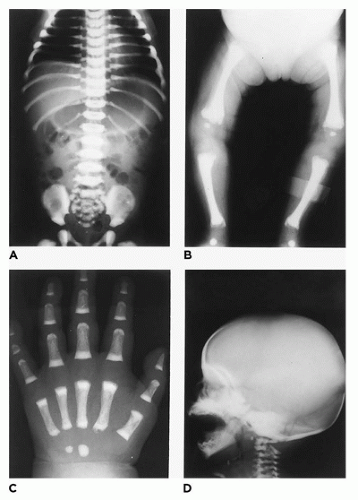
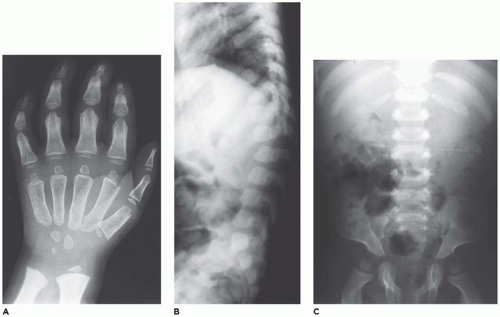
In population-based studies, both types of TD are almost as common as ACH (
144). Type 1 TD is characterized by
angulated or curved humeri and femora and craniosynostosis in 28% and mild cloverleaf skull in 3%, whereas type 2 is relatively straight femora, cloverleaf skull in 50%, and craniosynostosis in 90% (
Figure 28-4) (
178,
179). Angulated femora are also present in CD and OI type II (
178). Most infants die in the neonatal period of respiratory failure on the basis of severe secondary pulmonary hypoplasia as a consequence of the reduced volume of the thoracic cavity, which impedes normal lung growth (
180,
182). The lung/body weight ratio is low in contrast to the brain/body weight ratio (
180). The chondroosseous junction of the growth plate is substantially reduced in width with disorderly columnation of the chondrocytes as a reflection of the impaired FGFR3 signaling; there is fibrosis in place of regular chondroid ossification (
Figure 28-5) (
181,
182,
183). Other findings include the flattening of ossification centers (platyspondyly). Another aspect of the pathologic findings in TD is the range of neuropathologic abnormalities, which include overgrowth of the temporal lobe, hyperconvolution, and neuronal heterotopia (
187,
188).
Osteogenesis Imperfecta and Decreased Bone Density Group
Group 25 (ISDS classification) or OI is represented by two general categories: the so-called collagenous types or COL1A1-/2-related disorders with AD inheritance (OI types I, II, III, and IV) and the noncollagenous types with autosomal recessive inheritance (
Table 28-8) (
189,
190). It is estimated that 90% of all cases of OI have a defect in one of the type I collagen genes and the remainder are mutations in genes responsible for the synthesis of proteins that interact with collagen (
190,
191). With the failure of normal collagen type I synthesis by osteoblasts, the bones are less dense with the consequence
of structural fragility, which varies considerably in severity within the various types of OI (
192,
193). A severity index in a sense has been formulated and is based upon the type of OI (
194). The incidence of OI including all types is estimated at 1:10,000 to 20,000 live births; one case of OI type II is encountered for every two to five cases of TD in the perinatal-neonatal period (
195,
196).
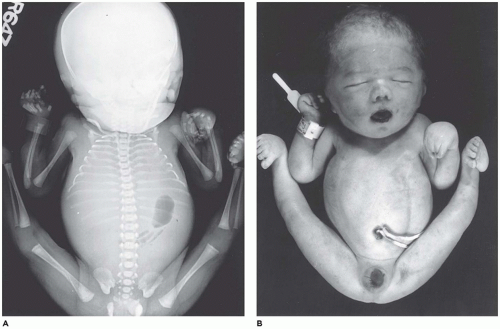
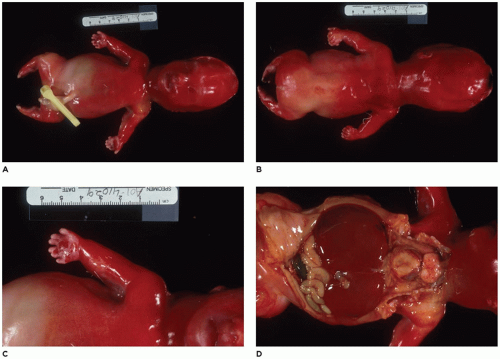
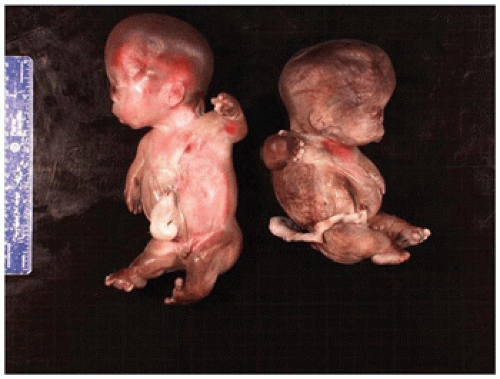
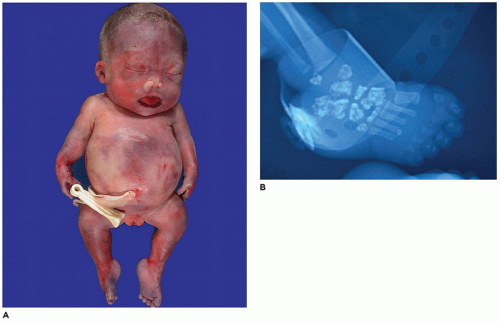
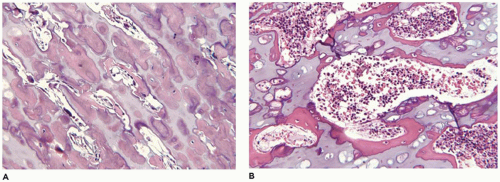
OI type II, known as the perinatal lethal type, is the manifestation of a new mutation in most cases and is characterized by severe osteopenia, blue sclera, short and bowed or angulated extremities, a diminutive thorax, and crumpled or collapsed long bones, especially the femora (
Figure 28-6A, B) (
195,
197). The cranium is soft and intracranial hemorrhage is not uncommon. Shortened, deformed extremities are also features of
achondrogenesis types 1A and 2, TD, and hypophosphatasia (HP). A small thoracic cavity with its deformities results in severe pulmonary hypoplasia with smaller than normal weight of lungs for gestational age and structural abnormalities of the thoracic cage. The bones are shortened, with multiple fractures with minimal normal callus formation, and multinodular chondroid masses are present that resemble an endosteal cartilaginous neoplasm or EC (
198). The cortex is quite attenuated, and the trabecular bone consists of delicate strands and is often disorganized, with an overall osteopenic appearance. The bone may appear hypercellular and the mosaic lines of osteoid seams are increased in number. The apparent hypercellularity is explained by a reduction in osteoid matrix secondary to defective type 1 collagen. The physis may be normal in many respects or may be disorganized (
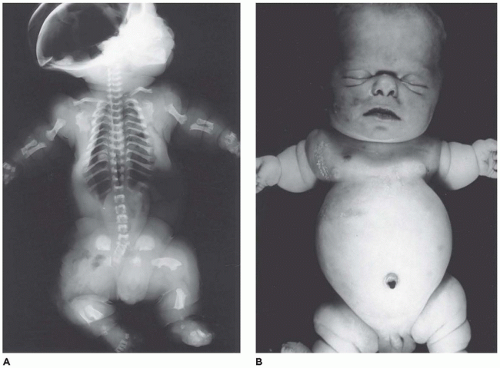
Figure 28-7A-D) (
199,
200). Chondrocyte columnation often appears normal, but osteoid forms directly on the cartilage without orderly endochondral ossification (
201,
202). These infants also exhibit neuropathologic changes, including perivenous microcalcifications and impaired neuroblastic-neuronal migration (
203).
OI type III, unlike type II, is usually not lethal in the perinatal period, but its severe phenotype is characterized by fractures and deformities of the lower extremities; these complications are present at birth and continue throughout life with the development of severe kyphoscoliosis (
194,
204). In one series, 25% of infants and children with OI have type III (
205). Lung infections with acute respiratory failure occur throughout the first decade of life because of the thoracic cage abnormalities (
206,
207). Marrow fibrosis and disorganized trabecular bone in OI type III can simulate fibrous dysplasia (FD). There are no specific histologic features to permit the differentiation of one type of OI from another (
208). Immature woven bone is prominent, and lamellar bone is poorly formed.
OI type V, unlike OI types I to IV, is not defined by mutations in collagen type I, but rather by a mutation in IFITM5-like protein, which interferes with the collagen triple helix and bone mineralization (
209,
210,
211). A moderate to severe phenotype and hyperplastic callus formation, especially in femoral fractures, can be mistaken for osteosarcoma (OS) (
212). Pseudoarthrosis, aortic and mitral valvular insufficiency, and aortic dissection are other manifestations (
213,
214). Multiple fractures in the absence of a prior diagnosis of OI can be mistaken for child abuse (
215). Rare examples of bone neoplasms and cysts have been reported in OI, including OS, chondrosarcoma (CS), ossifying fibroma (OF), and aneurysmal bone cyst (ABC) (
216,
217,
218).
OI and its definition have been challenging with the recognition of the autosomal recessive forms of the disease accounting for 10% of cases (
191,
193,
219,
220). There are presently eight genes that have been identified. Clinically, the recessive forms of OI present with bone fragility. One of these, Bruck syndrome, is associated with joint contractures (
221). The various recessive genes encode proteins, which are required for collagen transcriptional modification (
220,
222).
Defective Mineralization Group
Defective mineralization group (group 26) includes HP, an inborn error of metabolism associated with a deficiency of tissue nonspecific alkaline phosphatase with ALPL missense mutations in most cases (1p36.12) (
223,
224). The inheritance of the perinatal lethal and infantile forms of this disorder is AR with a prevalence of 1:100,000 births (
223). Approximately 2% to 4% of lethal GSDs are examples of perinatal HP. There are six clinical forms of HP, and these reflect the heterogeneity of the missense mutations on the ALPL gene (
225). There are some overlapping radiographic features among HP, OI types II and III, and achondrogenesis type 1A; however, these conditions can be differentiated from each other by radiographic analysis of the entire skeleton (
139). The histopathologic findings at the physis
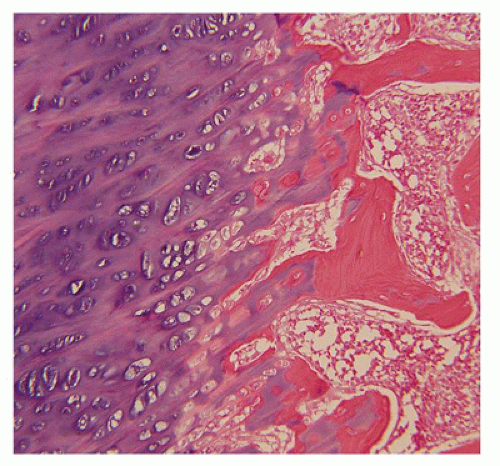
include a hypercellular, disordered osteochondral junction with cartilaginous overgrowth and minimal bone formation. Uncalcified osteoid with cores of cartilage is demonstrated in undecalcified sections. Some pathologic features of HP resemble those of rickets-osteomalacia.
Type 2 Collagen Group
Type 2 collagen group (group 2) comprises a phenotypically diverse category including lethal achondrogenesis type 2 (Langer-Saldino form), hypochondrogenesis, nonlethal spondyloepiphyseal metaphyseal dysplasia (Strudwick type), platyspondylic dysplasia (Torrance type), and Kniest dysplasia. In aggregate, this group of GSDs accounted for approximately 7% of cases in the Utah study (
144). Achondrogenesis type 2 (ACG 2) accounts for 5% to 7% of lethal GSDs whose inheritance is AD, and most cases are new mutations (
226). Hypochondrogenesis is a closely related entity, also with AD inheritance, with amino acid substitutions for glycine at different sites in type II procollagen. In addition to absent or minimal vertebral body ossification, cystic hygroma and/or hydrops fetalis are other features (
227). Severe pulmonary hypoplasia is the cause of death in the perinatal period in those cases that are carried to term. Complex congenital cardiovascular anomalies have been reported in hypochondrogenesis (
228). A papillomatous epidermal proliferation of the scalp with central ulceration has been reported in an infant with ACG 2 whose features are those of an epidermal nevus with possible aplasia cutis congenita (
229). Chondrocytes reside in enlarged lacunae, and the apparent hypercellularity is a consequence of diminished matrix in both ACG2 and hypochondrogenesis. Apparent “ballooning” of chondrocytes is described. Vascularity is increased in the reserve (resting) and proliferating zones of chondrocytes, and the columns of chondrocytes in the hypertrophic zone are irregular. Persistent central cores of cartilage are found within the bony trabeculae, as in HP and OP.
Short-Rib Dysplasia
SRD with or without polydactyly (group 9) includes chondroectodermal dysplasia (Ellis-van Creveld syndrome, EVC), which is generally compatible with life through the perinatal-neonatal period (
230,
231,
232). Several other entities in this group are Saldino-Noonan, Verma-Naumoff types 1 to 3, Majewski (type 2), Beemer (type 4) syndromes, and asphyxiating thoracic dysplasia (Jeune) (
233,
234,
235) are the other entities in this morphologic group (
Figure 28-8A to D) (
233,
234). Postaxial hexadactyly is a common feature of EVC. There is general recognition that group 9 dysplasias (ciliary chondrodysplasias) have mutations affecting the function of primary cilia in dynein motor, intraflagellar transport complexes, and basal body (
236,
237,
238,
239). Some minor histologic differences are noted in the physes in the various types of SRD, but in general, chondrocytic proliferation is diminished, as evidenced by a reduction in the thickness of the growth plate and disorganization of the columns of chondrocytes (
Figure 28-9) (
240,
241,
242,
243). Overlapping pathologic features are seen in EVC, ATD, and
renal-hepatic-pancreatic dysplasia (RHPD) of Ivemark; these conditions and others with similar RHPD-like changes are examples of primary ciliopathies (
244,
245,
246). Both EVC and Weyers acrodental dysostosis share mutations on the EVC2 gene (4p16); ectodermal dysplasia with enamel hypoplasia, hypodontia, and early eruption and exfoliation of teeth are seen on both disorders (
247,
248). Atrial septal or atrioventricular septal defects are present in 65% to 70% of EVC cases. Sensenbrenner syndrome (cranioectodermal dysplasia) with RHPD-like features is another related disorder (
249).
Severe Spondylodysplasias
Severe spondylodysplasias (SSDs) (group 14) are a category of platyspondylic lethal GSDs with overlapping phenotypic features with the FGFR3 group (group 1), including TD. Achondrogenesis type 1A (ACG1A) and Schneckenbecken dysplasias are two representative GSDs in this category (
250). Maternal polyhydramnios, fetal hydrops, and prematurity are accompanying complications. Absent or severely deficient ossification of the skull, vertebral bodies, and sacrum; shortened, beaded ribs; and small crescent-shaped ilia are some of skeletal anomalies in addition to shortened, bowed long bones. Microscopically, the growth plate in ACG1A is hypercellular, and the enlarged chondrocytes have a periodic acid-Schiff-positive, diastase-resistant inclusion within a cytoplasmic vacuole (
251). Mutations in TRIP11 (14q31-q32) in ACG1A result in apparent loss of function of the golgins GMAP-210 and reduced expression of COL10A1 (
252,
253). Schneckenbecken (snail pelvis) dysplasia, a PLSD with AR inheritance and mutation in SLC35DI, is also characterized by a snaillike pelvis and short limbs; there is some resemblance to TD (
Figure 28-10) (
254,
255,
256). Because of hypercellularity of the resting and proliferating zones of chondrocytes, the lacunar spaces are inapparent and the intercellular matrix is relatively inconspicuous. Reduced columnation of chondrocytes and hypervascularization are seen in the proliferating zones, and the chondrocytes have uniformly centralized nuclei. The classification points out that PLSDs are found in group 1 (TD), group 2 (ACG 2 and Torrance dysplasia), group 3 (fibrochondrogenesis), group 4 (ACG1B), and TRPV4 group (metatropic dysplasia) (
138).
Spondyloepi(meta)physeal Dysplasias
Group 13 dysplasias account for 5% or fewer of skeletal dysplasias (
144). This category of GSDs has undergone substantial revision, especially in regard to metatropic dysplasia, which has been assigned to the TRPV4 group (group 8). Schimke immunoosseous dysplasia is an AR disorder with biallelic missense mutations in the SMARCA1 gene (2q34-q35) (
257,
258,
259). There is one case per 1 to 3 million live births (
260). A range in clinical severity with death is seen within the first 5 years of life or clinical onset in later childhood with the development of the nephrotic syndrome whose pathologic findings are the collapsing variants of focal segmental glomerulosclerosis (
261). There is in utero growth restriction. T-cell immunodeficiency is complicated by autoimmune thyroiditis, infections, and neoplasms like lymphoma, poorly differentiated carcinoma, and OS (
262,
263,
264). Accelerated atherosclerosis with complications of cerebral ischemia is thought to be a consequence of reduced elastogenesis (
265). The histologic findings in the physis are a hypocellular, attenuated growth plate. There is little evidence of chondrocyte enlargement and poorly defined chondrocyte columnation.
TRPV4-Associated Disorders
Group 8 includes those skeletal dysplasias with AD mutations in the TRPV4 gene (12q24.1) (
266). This clinical category includes GSDs and neuromuscular disorders. Brachydactyly is a consistent feature of TRPV4—GSDs and short stature with progressive spinal deformity in the severe forms. There are individuals with a hybrid phenotype of peripheral neuropathy and skeletal dysplasia, including fetal akinesia (
267). The TRPV4 gene encodes a protein that is thought to form a cation-conductive pore or calcium channel (
268). Metatropic dysplasia (MD) has a spectrum of clinical severity from the perinatal and early childhood lethal forms to the mild to severe nonlethal forms (
269,
270). The phenotype of MD is as heterogenous as its clinical manifestations (
271). Short extremities, bowing of long bones, craniosacral anomalies, decreased bone density, and flattened vertebral bodies (platyspondyly) are the manifestations of irregular enchondral ossification. Short ribs and a diminutive thorax correlate with pulmonary hypoplasia in the lethal form. A caudal appendage has been reported in lethal MD (
272) (
Figure 28-11A, B). A disorganized primary spongiosa with irregular trabeculae, diminished ossification of epiphyseal cartilage in tubular bones, and poor chondroosseous columnation and hypercellular cartilage are some of the microscopic findings.
Sulfation Disorders
Sulfation disorders (
SDs) (group 4) were present in 2% of infants with GSDs in the Utah population study (
144). These AR disorders include four with mutations in the DTDST gene (5q 32-33): achondrogenesis type 1B (ACG1B) (
Figure 28-12A, B), atelosteogenesis type 2 (AO2), diastrophic
dysplasia (DD), and multiple epiphyseal dysplasia, recessiva (
273). There is considerable phenotypic overlap between AO2 and DD (
274). The pathologic features of DD and AO are similar: an attenuated growth plate, irregular clumping of chondrocytes in the resting zone, and foci of myxoid degeneration, not necessarily confined to the resting zone. One of the hallmark features is a dense rim of collagenous matrix around each lacuna (
275,
276). Giant chondrocytes may be found as well. ACG1B, in addition to severe micromelia with marked shortening of the femora and humeri, is characterized by minimal or absent ossification of the vertebral bodies and malformed tibiae and fibulae (
Figure 28-13) (
277).
Filamin Group and Related Disorders
Group 7 is defined by the presence of mutations in the FLNA gene (Xq28) that encodes filamin A and includes the following X-linked dominant dysplasias: frontometaphyseal dysplasia, Melnick-Needles osteodysplasia, and otopalatodigital syndromes types 1 and 2 (
278). Mutations in FLNB gene (3p14.3) are both AD and AR and include the following: AO1, AO3, Larsen syndrome, spondylocarpotarsal syndrome, Piepkorn, and boomerang dysplasias (the latter two have been included with AO1) (
138,
279,
280,
281). The filamins are actin binding that stabilize the structure of actin and link them to the cell membrane (
282). The filamin A-associated disorders are diverse from the skeletal dysplasia to periventricular nodular heterotopia to severe congenital lung disease with cysts and pulmonary vascular hypertensive changes (
283,
284,
285). Otopalatodigital syndrome type 2 is a potentially lethal disorder with thoracic and pulmonary hypoplasia. Other defects include hypomineralized calvarium, poorly formed small bones of the hands and feet, septal and right ventricular outflow tract defects, omphalocele, and genitourinary tract anomalies. Boomerang dysplasia and AO1 are lethal disorders due to the small thorax and hypoplastic lungs (
286,
287). Multinucleated and giant chondrocytes may be seen in a focally hypocellular reserve or resting zone. Similar giant chondrocytes have been seen in Piepkorn dysplasia, which may be allelic to boomerang dysplasia (
288). Near-complete
absence of ossification and mineralization are seen in boomerang dysplasia. Marked disorganization in the columns of chondrocytes is the histologic features in boomerang dysplasia.
Chondrodysplasia Group
Chondrodysplasia (
CDP)
group (group 21) comprised 4% of GSDs in the Utah study (
144). There are also acquired causes of CDP-like changes in children unrelated to the GSDs with similar punctate-stippled calcifications in the epiphyses and around the spine; examples are prenatal exposure to warfarin and neonatal lupus erythematosus (
289). In terms of pathogenesis, CDP can be classified into inborn errors of cholesterol biosynthesis, peroxisomal biogenesis disorders, disruption of vitamin K metabolism, and chromosomal abnormalities (
290,
291). Rhizomelic CDP type I and Zellweger syndrome, both AR peroxisomal disorders, are lethal in most cases (
292). Conradi-Hünermann-Happle (CHH) syndrome, with X-linked dominant inheritance (Xp11.23-p11.22), may be lethal in the affected neonate (
Figure 28-14A, B) (
293,
294). One of the characteristic and accessible pathologic findings in CHH syndrome is lamellar orthokeratosis and dystrophic calcifications in keratotic plugs in a skin biopsy as the features of the linear ichthyosiform lesions (
295). Rhizomelic CDP is represented by three AR disorders: peroxisomal CDP1 (PEX7 on 6q22-q24), CDP2 (DHAPAT on 1q42), and CDP3 (AGPS on 2q31) (
296). The incidence is 1:100,000 live births. Severe shortening of proximal long bones (rhizomelia), cataracts, dysmorphic facies, and severe growth abnormalities are the various clinical features. Approximately 50% or more of children do not survive beyond the age of 6 years. The cause of death is usually respiratory in nature after multiple respiratory tract infections. Dystrophic calcifications in the region of an otherwise unremarkable growth plate and cystic myxoid degeneration in the subarticular cartilage are the principal histologic features in rhizomelic CDP (
297). In other conditions, association with stippled calcifications, dystrophic calcifications, and degenerative changes in the chondroid matrix are the microscopic findings (
Figure 28-15). OI-like features are present in the lethal Astley-Kendall syndrome as one of the overlap syndromes, which in this case is classified with the group 21 disorders (
298).
Bent Bone Dysplasia
Bent bone dysplasias (
BBDs) (group 18) are characterized by short limb dysplasia and bowing of the long bones of the lower extremity (
299,
300). However, the presence of “bent bones”
may be seen at birth in several other GSDs (
138). The Utah study had six (4%) cases of BBD and five were examples of CD (
144). CD has a reported incidence of 1:200,000 births and accounts for approximately 4% to 6% of all lethal skeletal dysplasias (
301,
302). Mutations in the SOX9 gene (17q24-q25) are the molecular genetic defect in this AD disorder (
303). The role of SOX9 is multifold in development and in the context of this discussion—chondrogenesis with the activation of multiple cartilage-specific genes (
304). Anterior bowing of the long bones of the lower extremities, hypoplastic thorax with secondary pulmonary hypoplasia, and craniofacial anomalies are the phenotypic abnormalities in addition to the characteristic sexual anomalies with so-called sex reversal; it is estimated that gonadal dysgenesis with male to female sex reversal is present in 75% of CD cases (
305). Abnormalities of both müllerian and wolffian duct structures and incomplete ovarian or testicular development are other findings; gonadoblastoma may be seen in the dysgenetic testis (
306,
307). The physes of the long bones exhibit minimal histologic abnormalities; however, some alterations in the proliferative and hypertrophic zones of chondrocytes may be seen. In place of normal cortical bone, immature woven bone, osteoclastic activity, and vascularized intraosseous spaces are the microscopic features. Stuve-Wiedemann syndrome (SWS), another group 18 GSD, is a severe AR disorder with mutations in the LIFR gene (5p13.1) (
308,
309). Cortical thickening is present in bowed long bones with flared metaphyses and progressive decalcification. Dysautonomic hyperthermia, respiratory complications on the basis of aspiration pneumonitis, pulmonary artery hypertension, and cutaneous infections all contribute to death by 2 years of age (
310,
311). These children like those with CD have obstructive airway problems (
312). BBD is also seen in FGFR2 mutation (
313,
314).
Neonatal Osteosclerotic Dysplasias
Neonatal osteosclerotic dysplasias (NOD) and increased bone density (IBD) group are designated as group 22 and group 23, respectively. Group 22 GSDs comprise five disorders including Caffey disease (infantile cortical hyperostosis), which is primarily an inflammatory and reactive condition arising in the diaphyseal region of long bones, but also in the mandible and clavicle in infants. There is fever and soft tissue swelling overlying the affected bone (
315). The mandible is most commonly involved in 70% to 90% of cases (
316). A mutation in the COL1A1 gene is present in the prenatal or infantile forms of Caffey disease but is uncommon in the later presenting form (
317,
318,
319). Blomstrand dysplasia is an AR lethal neonatal disorder, which is characterized by generalized osteosclerosis and advanced skeletal maturation (
320). Group 22 also includes desmosclerosis and Raine dysplasia. Inactivating recessive mutations in the PTH-related peptide type I receptor gene PTH PTH1 (3p22-21.1) are present in Blomstrand dysplasia (
321,
322). This same gene is mutated in Jansen chondrodysplasia. In addition to very short stature, the limbs are micromelic with accelerated ossification of virtually the entire skeleton. Like the other lethal skeletal dysplasias, the thorax is short and narrow. Early ossification of the epiphyseal center, a reduction in the epiphyseal cartilage, irregularity of the transformation zone, subperiosteal ossification, and cortical hyperostosis are some of the histologic findings.
Increased Bone Density: Osteopetrosis
IBD or OSP (group 23) is characterized by osseous hyperdensity without alteration in the shape of the bones. The basic defect occurs in the osteoclast in either development or function (
323,
324,
325). Based upon the particular mutation, OSP is divided into the “osteoclast rich” whose mutations affect function and “osteoclast poor.” The severe, neonatal forms with AR inheritance are seen in 1:250,000 births and are associated with several mutations (
326,
327,
328). Loss-of-function mutations have been identified in five genes: TCIRG1 (11q13), CLCN7 (16p13), OSTM1(6q21), RANKL (TNFSF11, 13q14), and RANK (TNFRSF11A, 18q22.1) (
325,
329,
330). Among these mutations in the AR OSP, the TCIRG1 mutation is detected in 50% to 60% and CLCN7 mutations account for 20% of cases and CLCN7 mutation in 20% to 25% of cases (
324,
325). Leukopenia and hepatosplenomegaly are the consequences of bony overgrowth of the marrow space with bone marrow failure, phthisic anemia, and organomegaly on the basis of extramedullary hematopoiesis. Early death may result from pathologic fractures, hydrocephalus, or anemia; the pathologic fractures are typically transverse breaks in the long bones. Without bone marrow or stem cell transplantation, survival beyond infancy is rare. Another expression of AR OSP is renal tubular acidosis (RTA) with cerebral calcifications; there are mutations in the carbonic anhydrase II gene (8q21.2). OSP-RTA is usually detected in the first 2 years of life with growth failure, mental retardation, visual and auditory deficiencies, pathologic fractures, and metabolic acidosis (
325). Cerebral calcifications are detectable after 18 months of age. Cortical bone thickening is present in all forms of OSP, but in the most severe cases, the marrow cavity is obliterated and the corticomedullary demarcation is lost (
Figure 28-16A to D). Osteosclerosis may be uniform or alternating, as seen in the vertebrae, where transverse striations of alternating lucent and dense bone formation with the so-called ruggerjersey spine. Lucency of the central portions of bones may convey the appearance of “bone within bone.” Coxa vara and lateral bowing of the long bones are common findings, and rachitic features may be observed in infants. The microscopic hallmark is the persistence of calcified cartilage, surrounded by dense woven or lamellar bone of endochondral origin (
Figure 28-17A, B) (
331). The zone of proliferating cartilage is often extremely wide at sites of active endochondral ossification in infants, which reflects the failure in remodeling of mineralized cartilage and bone. Woven bone persists in the absence of lamellar bone formation. The number of osteoclasts in a bone biopsy may depend on the sampling or the osteoclast-rich or osteoclast-poor status of the particular type. Howship lacunae are often difficult to identify. Osteoblasts are generally present in normal numbers, but they often appear flattened and inactive. The marrow space is more or less obliterated by woven rather than lamellar bone, which is thought to account for the extreme fragility of the bone
despite its increased density. Ultrastructurally, the osteoclasts adjacent to the bone surface may lack the ruffled membrane that is necessary for bony resorption, which is impaired in all forms of OSP. AD OSP has mutations in CLCN7 and does not present until late childhood or adolescence with fractures, scoliosis, osteoarthropathy, and osteomyelitis of the mandible (
325). The previous type of AD OSP with an LRP5 mutation is no longer regarded as a classic form of OSP (
332).
Lysosomal Storage Disease
Lysosomal storage diseases (
LSDs) with skeletal involvement or dysostosis multiplex group (group 27) comprise a family of heritable metabolic diseases with a defect in a specific acid hydrolase or enzyme activator whose functional and morphologic consequences are an accumulation or storage of the catabolic product at the blocked biochemical step. There are approximately 50 LSDs recognized, and the storage products include mucopolysaccharides, glycoproteins, amino acids, and lipids (
333,
334). Currently, 22 disorders with similar radiographic abnormalities are included in the ISDS classification (
138). The incidence of LSDs is approximately 1:1500 to 8000 live births (
335,
336). Hypoplastic iliac bones with pseudoenlargement of the acetabula, pointed proximal metacarpals, defective development of the anterosuperior portions of the vertebral bodies at the thoracolumbar junctions, and widened ribs that taper near the vertebral margins are among the more consistent skeletal abnormalities among the LSDs with some differences among the specific types (
Figure 28-18A to C) (
337,
338). The changes in the tubular bones, which are more pronounced in the upper extremities, include diaphyseal and metaphyseal expansion, delayed epiphyseal ossification, and osteopenia. Other changes include macrocrania, coxa valga, small carpal bones with V-shaped deformities of the distal radius and ulna, cardiomegaly, and hepatosplenomegaly. A consistent histologic finding in various parenchymal and mesenchymal cells, from hepatocytes to chondrocytes, is cellular enlargement, which
reflects the presence of numerous membrane-bound vacuoles representing distended lysosomes that are clear or contain finely granular material (
339,
340,
341) (see
Chapter 5).
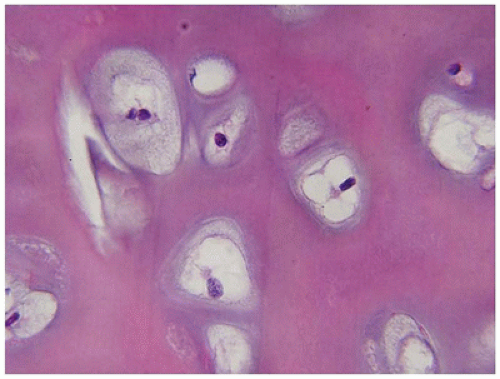
Mucopolysaccharidoses (
MPSs) are the most familiar and common LSDs. There are seven distinct eponymic types of MPS with a total of 11 enzymatic defects (
342,
343). The overall incidence of the MPSs ranges from 1:100,000 to 600,000 live births with AR inheritance in all but MPS/H with X-linked recessive inheritance. A specific chromosomal defect has been identified in all of the MPSs. The natural history of the MPSs depends upon the severity of the CNS involvement and the development of cardiorespiratory complications (
344,
345,
346,
347). Shortened stature and progressive skeletal deformities are indicative of growth plate and bony abnormalities with subluxation of joints and progressive kyphoscoliosis, as in MPS type 7 (Sly syndrome). The growth plate is reduced in thickness and is disorganized in appearance. The prominence of enlarged, vacuolated chondrocytes varies somewhat among the types of MPS (
Figure 28-19). Abrupt calcification of the cartilage without the formation of primary trabeculae is another feature of the growth plate in several of these disorders, including MPS type 1H (Hurler syndrome), MPS type IH/1S (Hurler-Scheie syndrome), and MPS type 4A (Morquio syndrome) (
348). In addition to abnormalities in the axial and appendicular skeleton, histologic changes in the temporal bone have been correlated with deafness in MPS type 1H, type 1 IS (Scheie syndrome), MPS type 1H/1S, and MPS type 2 (Hunter syndrome).
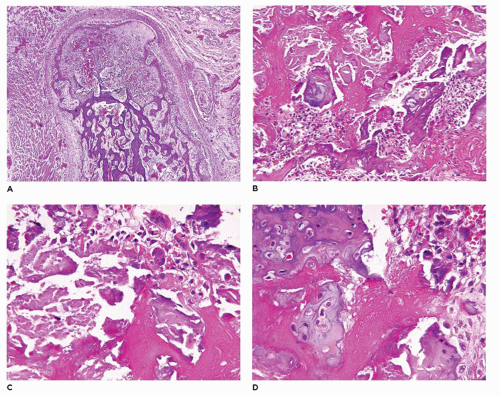
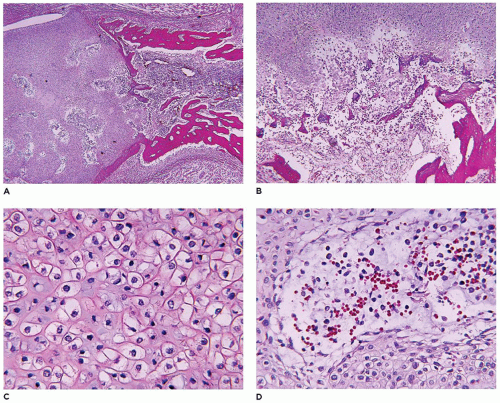
Gaucher disease (GD), the most common LSD, occurs in 1:100,000 individuals in the general population, but 1:800 to 1000 Ashkenazi Jews (
349). Type 1 GD accounts for 99% of cases, which becomes symptomatic in adolescence and early
adulthood. Bone changes in the distal femur and/or proximal tibia are present in 70% to 100% with type 1 GD (so-called adult) or type 3 (juvenile) GD (
350,
351,
352). In addition to the Erlenmeyer flask-like deformities, osteopenia with pathologic fracture, bone infarcts, and osteomyelitis are the other skeletal complications. Macrophages with glucocerebrosides have a granular eosinophilic and fibrillary appearance.
Several other LSDs distinct from MPS also have dysostosis multiplex features, including mucolipidosis 2 (I-cell disease) and mucolipidosis 3 (pseudo-Hurler polydystrophy). Yet another category of metabolic disorders includes those in which glycoprotein degradation and structure are defective: fucosidosis, α-mannosidosis, β-mannosidosis, sialidosis, aspartylglycosaminuria, sialic acid storage disease, multiple sulfatase deficiency, and galactosialidosis (
353,
354,
355). Carpal tunnel syndrome is a complication in both the MPSs and mucolipidoses secondary to the accumulation of material in swollen fibroblasts and the presence of foamy histiocytes (
356).
Melorheostosis With and Without Osteopoikilosis, Pyknodysostosis, and Osteopathia Striata
Melorheostosis with and without osteopoikilosis, pyknodysostosis, and osteopathia striata is an example of a group 23 disorder (
138). The estimated prevalence of melorheostosis is 1:1,000,000 individuals (
357,
358). The overwhelming majority of cases are sporadic in occurrence, but isolated examples have been reported in association with osteopoikilosis with mutations in the LEM domain containing 3 gene (12q14) (
156). Mutations in this same gene are found in Buschke-Ollendorff syndrome. The diagnosis is rarely made in infancy, but 40% to 50% of cases are discovered before the age of 20 years (
359). Any bone or bones may be affected; however, involvement is frequently unilateral, with one or more hyperostotic long bones, usually in the lower extremity, with so-called flowing hyperostosis (
360). The fibrosing component in the contiguous soft tissues has fibromatosis-like or atypical decubitus fibroplasialike histologic features, which result in contractures, a cause of substantial morbidity in this disorder (
361). Fibrofatty and myositis ossificans-like lesions have also been observed. Unlike the marrow space in OP, the marrow space remains intact, but the cortex is thickened and dense, with a paucity of haversian canals. Mosaic lines may be prominent. Osteoclastic activity is inapparent, whereas osteoblasts are present, but not in appreciable numbers. Endochondral ossification extends well into the zone of articular cartilage. Osteopoikilosis is an AD disorder in which bone islands form at the ends of a bone and in the vicinity of the metaphysic. Histologically, the foci are identified as rounded expansions of hyperdense bone with some mosaic lines. Multiple dermal fibrous papules in association with osteopoikilosis are the features of Buschke-Ollendorff syndrome.
Metaphyseal Dysplasias
Metaphyseal dysplasias (
MPD) (group 11) comprise a genetically heterogenous group of disorders, which are characterized by a failure in enchondral bone growth and remodeling of the end of the long bones with the development of Erlenmeyer flask-like deformity of the metaphysis with an increased diameter (
362,
363). The distal femur and proximal tibia are the most frequently affected sites. Similar deformities are seen in GD and other GSDs, which are not classified among the group 11 diseases. Shwachman-Diamond syndrome, one of the inherited bone marrow failure syndromes, in addition to FA, Diamond-Blackfan anemia, dyskeratosis congenita, and Kostmann severe congenital neutropenia, is an AR disorder with an incidence of 1:75,000 births; 90% of cases have mutations in the SBDS gene (7q11) (
364,
365). Metaphyseal dysplasia of the femoral head is present in 50% of cases, but abnormalities in ribs (shortened with flared ends) can result in a hypoplastic thorax with lethal consequences in the neonatal period (
366). There is an apparent failure in the formation of the zone of hypertrophic cartilage; however, a case has been reported with features of spondylometaphyseal dysplasia in a neonate with a SBDS gene mutation in which the hypertrophic zone was hypercellular with minimal matrix extending into the metaphysis (
367). Cartilage-hair hypoplasia (CHH, McKusick type) is one of four skeletal dysplasias with mutations in the RMRP gene (9p21-p13) (
368,
369). The incidence is 1:23,000 live births (
370). In addition to short stature, these children have ectodermal dysplasia and T-cell immunodeficiency (
371). Granulomatous inflammation of the skin may occur in infancy (
372,
373). Another type of MPD is the Schmid type with a mutation in COL10A1 gene (
155,
374).
Genetic Inflammatory Rheumatoid-Like Osteoarthropathies
Genetic inflammatory rheumatoid-like osteoarthropathies (
GIRLOs) (group 31) include several autoinflammatory disorders with both AR inheritance and AD inheritance (
138). The pathogenesis of these hereditary autoinflammatory disorders is an apparent disruption in the linkage of IL-1 function and the regulation of the innate immune response (
375,
376). Mutations in two IL-1-regulating genes, NLRP3 and IL1RN, are responsible for cryopyrin-associated periodic syndromes (CAPS) and deficiency of IL-1 receptor antagonist (DIRA). CAPS is a spectrum disorder whose earliest and severest manifestations are present in the neonatalonset multisystem inflammatory disease (
377,
378). There is overgrowth in the region of the growth plate with exostosislike features, defects in limb lengths, and contractures. A skin biopsy shows a neutrophilic infiltrate of an urticarial or neutrophilic dermatosis (pyoderma gangrenosum). In addition to growth retardation, there is diffuse osteopenia. Muckle-Wells syndrome is a less severe form of NOMID/CINCA spectrum (
379). Chronic recurrent multifocal osteomyelitis (CRMO) and nonbacterial acute osteitis are associated with mutations in IL1RN or LPIN2 (Majeed syndrome) (
380,
381). Other mutations in CRMO include GALNTS and RAGS. The median age at diagnosis is 10 years ,and the presentation is the development of osteolytic bone lesions with the pathologic features of acute to subacute to chronic osteomyelitis; however, the lesions are sterile for microorganisms. Progressive pseudorheumatoid dysplasia develops in children between 3 and 6 years and has a mutation in the WISP3
gene (
382). There are minimal signs of inflammation unlike the other disorders in this group. Growth abnormalities are present, and multiple large and small joints are involved with a particular early predilection for the hip joints where there is enlargement of the femoral heads.
Disorganized Development of Skeletal Component Group
Disorganized development of skeletal component group (group 29) constitutes several tumefactive lesions of bone, some of which are familiar to pathologists including polyostotic FD, multiple osteochondromas (OCs), or multiple hereditary exostoses, cherubism or multiple giant-cell reparative granulomas (GCRGs), and enchondromatosis with or without hemangiomas (
138). These various tumor and tumorlike lesions are discussed in the subsequent section on neoplasms.