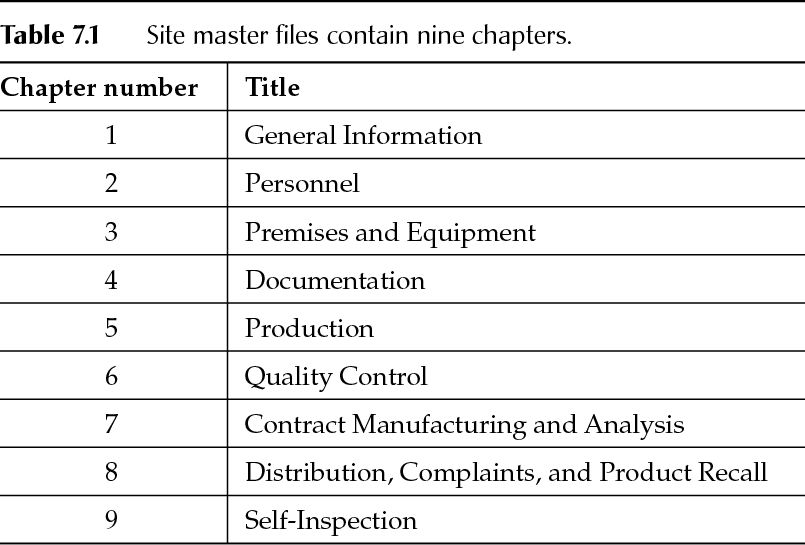
Content of Site Master Files
Chapter 1. General Information
• Name, address, and company description (C.1.1): Includes a brief description of the company, relation to other sites, and any information relevant to understanding the manufacturing operations.
• Licensed pharmaceutical manufacturing activities (C.1.2): Provides the relevant document issued by a national authority, and, if applicable, the period of validity for the manufacturing license. This section includes any conditions/restrictions of the manufacturing license and, where applicable, the drug master file (DMF) for each product.
• Description of other pharmaceutical and nonpharmaceutical activities (C.1.3).
• Name and address of the site (C.1.4): Includes the name of the site, the complete street address and postal address, the telephone number, fax number, and e-mail address of a contact person, and a 24-hour contact telephone number.
• Products manufactured at the site (C.1.5): Lists the types of products manufactured at the site and provides information about specific toxic or hazardous substances handled, including their manufacturing. This section provides information on whether the products are manufactured in a dedicated facility or on a campaign basis and states whether both human and veterinary products are manufactured on the site. For contract manufacturing or analytical sites, this section defines whether the firm is the contract giver or acceptor.
• Description of the site (C.1.6): Provides the location and immediate environment, and the size of the site, types of buildings, and their ages.
• Number of employees engaged in production, quality control, storage, and distribution (C.1.7): Provides the total number of employees and the number of employees engaged in production, quality control (QC), quality assurance (QA), storage and distribution, and technical and engineering support services.
• Listing and description of outside scientific, analytical, or other technical assistance in relation to manufacture and analysis (C.1.8): Provides the company name, address, telephone number, and fax number of each outside contractor, and includes a brief outline (not more than 100 words) of the activity being undertaken. Also includes the establishment license number, where applicable.
• Description of the quality management system (QMS) (C.1.9): Provides a short description (750 words or three A4 pages) of the QMS of the firm responsible for manufacture. Describes the quality policy and the responsibilities of the QA function, including audit programs, approval and starting, primary packaging material suppliers, batch release, and document control. The responsibilities specific to the head of quality, QA manager, and QC manager are included. Describes the elements of the QA system (for example, organizational structure, responsibilities, procedures, and processes, specifications, test methods, and other quality-related data collection). It is important to show that quality management is independent from production management. Briefly describes the self-inspections or audits undertaken by external organizations. A cross-reference to Chapter 9 can be added for additional details regarding the self-inspection program. Describes how quality system results are reviewed to demonstrate the adequacy of the quality system in relation to the objective. Identifies the standards used to assess or audit the company’s quality system, or used by the company to assess suppliers, such as ISO 9001, 21 CFR 211, and EU GMP. When suppliers of critical materials and packing materials are assessed, this section details how this is done, and describes the supplier selection process.
Chapter 2. Personnel
• Organization charts (C.2.1): Organization charts define the overall organizational structure for QA, production, and QC, with detailed charts for each area, depicting senior managers and supervisors only. As this information also is included in section 2.1, a cross-reference to that section can be used.
• Qualifications, experience, and responsibilities of key personnel (C.2.2): Includes brief details of academic qualifications, work-related qualifications, and years of relevant experience since qualifying.
• Training program (C.2.3): Describes how training needs are identified and by whom. Gives details of training relative to GMP requirements, including new employee training and continuous GMP training. Describes job-related skills training and in-house and external training, and how practical experience is gained. Explains how retraining needs are identified and details how training is recorded and records are maintained.
• Health requirements for production personnel (C.2.4): Describes who is responsible for checking the health of employees. Describes requirements for preemployment medical examinations and how employees are checked depending on the nature of their work. Describes the system for reporting sickness or contact with sick people before working in a critical area. Provides information on any system of reporting back to work after illness, and the requirements for personnel who work in sterile areas and any additional monitoring to which they may be subjected.
• Personnel hygiene and clothing requirements (C.2.5): States the washing, changing, and rest areas and the clothing/gowning requirements for the production areas. Gives instructions on how clothing should be used and when it should be changed. Describes gowning room/change room procedures. States the rules on eating, drinking, smoking, and use of chewing gum or tobacco. It is helpful to inspectors to include references to the applicable procedures for hygiene and clothing requirements.
Chapter 3. Premises
• Site plan and manufacturing areas (C.3.1): Provides a site plan highlighting the production areas, and a simple plan of each production area with indication of scale. For sterile product areas, indicates room and area classification(s) and pressure differentials between adjoining areas of different classifications.
• Nature of construction and finishes (C.3.2): A narrative format is preferred for this section. To reduce the length of the narrative for a large, complex plant, the details should be limited to critical areas, including processing and packaging areas. Describes wall construction, nature of finishes, floors, ceilings, doors and windows, lighting, piping, and drainage system(s).
• Brief description of ventilation system (C.3.3): To reduce the length of the narrative, schematic drawings should be used where possible. Details should be given for critical areas with potential risks of airborne contamination, including sterile product areas and areas for processing powders, granulation, and tableting. For manufacture of sterile or aseptic products, classification of the rooms used for production should be mentioned. A summary of the results of the most recent qualification/requalification should also be included. The MHRA guidance specifies that room classification should be given in accordance with the grading system outlined in the EC/PIC Guide to GMP. Outlines the heating, ventilation, and air conditioning (HVAC) system, areas with different classes of air, pressure differential principles to prevent cross-contamination, dedicated air handling units (AHUs), and forced ventilation systems. The limits for changing the filters should be given. If a drop-off point (DOP) is introduced, the point must be shown.
• Special areas for the handling of highly toxic, hazardous, and sensitizing materials (C.3.4): This section is similar to section C.3.1, and includes information such as whether materials are handled only in the QC laboratory, whether solids or waste are neutralized and collected in separate containers, and whether extracted gases from the fume hood are neutralized and scrubbed before they are allowed to exit into the external environment.
• Description of water and steam systems (C.3.5): This section provides a schematic drawing back to the main city supply system or source of the raw water, and the capacity of the system (maximum quantity produced per hour). Construction materials of the vessels and distribution system (pipework) are provided, along with specification of any filters in the system. If water is stored and circulated, the temperature at the point of return is provided, and the specifications of the water produced (that is, chemical, conductivity, and microbiological descriptions) are provided. The sampling points and frequency of sampling should be included. If a steam system is needed, the same sort of information must be provided for the system.
• Description of planned preventive maintenance program and recording system (C.3.6): “Maintenance” is done by the manufacturer and “servicing” by an outside contractor. This section describes the planned preventive maintenance program, including written procedures and suitable reporting forms for maintenance and servicing.
• Brief description of major production and control laboratories equipment (C.3.7): This section provides a list of equipment. Although the make and model numbers of the equipment are not required, several points should be addressed, including appropriateness and validity of construction material and ease of cleaning. For specific pieces of machinery (for example, a rotary tablet press), only a general description is required. If the equipment has additional devices, however, these devices should be recorded (for example, automatic weighing machines with printer, a labeler incorporating a bar code reader for the label). Further information is required for QC laboratory and microbiology laboratory and for computers and microprocessors in the manufacturing facility.
• Maintenance (description of planned preventive maintenance program and recording system) (C.3.8): Identifies who is responsible for maintenance and servicing and whether there are written procedures and contractual details for servicing. Maintenance routines that could affect product quality must be identified, and annual planned preventive maintenance programs for equipment must be in accordance with machine/instrument manufacturers’ requirements. Documentation must be provided that records are kept on the type and frequency of service or check, details of service repairs and modifications, and reports provided to the users.
• Qualification, validation, and calibration, including recording system and arrangements for computerized systems validation (C.3.9): This section briefly describes the company’s general policy and protocols for qualification, prospective validation, and retrospective validation, and describes and outlines the essential steps of the manufacturing process validation and cleaning validation. Any revalidation policy must be described. An outline of process validation may be given here or cross-referenced to production paragraph 5.4. A description of the system for the release for sale or supply of development and validation batches should be provided, as well as the arrangements for computer validation, including software validation. The equipment calibration policy and how calibration records are kept should be included in this section.
• Availability of written specifications and procedures for cleaning manufacturing areas and equipment (C.3.10): This section summarizes the cleaning strategy, schedules, and documentation for manufacturing areas and equipment. If cleaning agents are changed periodically, it should be noted, along with any validation of cleaning processes, including the cleaning methods (and their frequency) for the water supply system, air handling system, and dust extraction system.
Chapter 4. Documentation
• Arrangements for the preparation, revision, and distribution of necessary documentation for manufacture (C.4.1): This section includes a description of the documentation system and who is responsible for the preparation, revision, and distribution of documents. The storage place for the master documents is described, as well as type of documents stored (that is, product/process specifications, raw material specifications, packaging component specifications, standard process instructions including packaging, batch records including packaging, analytical methods, and QA release procedures). Information on how documentation is prepared and controlled and retention policy is provided, as well as the arrangements for any electronic or microfilmed records.
• Other documentation related to product quality (C.4.2): This section includes any other relevant documentation related to product quality that is not described elsewhere (for example, microbiological controls on air and water). The section should list and briefly explain the use of any additional standard documentation used routinely, including documentation for equipment specifications, specifications for disposables, standard operating procedures (SOPs), QC procedures, training procedures, computer program specifications, documentation control of process deviations, other validation documents, and reconciliation of batches of raw materials, bulk product, and major packing components (for example, product-contact and printed materials).
Chapter 5. Production
• Brief description of production operations using, wherever possible, flow sheets and charts specifying important parameters (C.5.1): This section should describe the operations capable of being done at the site with the existing facilities, and specify the categories of medicinal products produced. In facilities where only packaging is undertaken, it is necessary only to provide a brief description of labeling and filling and the nature of the containers used (for example, tamper-evident glass containers). More information is required in this section if cytotoxic or radioactive substances are handled. Flowcharts are useful to describe the production operations; technical details are not required.
• Arrangements for the handling of starting materials, packaging materials, bulk and finished products, including sampling, quarantine, release, and storage (C.5.2): Purchasing procedures (approval of suppliers of active pharmaceutical ingredients, excipients, and primary packing materials), and material requisition procedures from stores to manufacturing plant and vice versa, including sampling, quarantine, release, and storage, are all described in this section. Identification of supplier’s lot number with the company’s lot number is provided, along with sampling plans. Other details to be included are status labeling (for example, by using labels or by computer), issue of materials to manufacture and package, control of weighing, checking methods, and how materials being used for manufacture are identified and confirmed. Information for the control of bulk manufacturing (that is, records of key parameters and in-process checks) and packing (that is, release of bulk, semifinished product, and packing materials, along with in-process checks) is provided.
• Arrangements for the handling of rejected materials and products (C.5.3): Information must be given to show that rejected materials are clearly labeled and that they are stored in a dedicated area. This section also describes the arrangements for sentencing the materials and their disposal.
• Brief description of general policy for process validation (C.5.4): An outline of process validation protocol only is required. An outline may be given here or cross-referenced to Chapter 3 (3.9).
Chapter 6. Quality Control
Chapter 6 provides a description of the QC system and activities, particularly the procedures for the release of finished products. The information includes a description of the analytical testing, packaging, component testing, and microbiological testing, and outlines the arrangements for the preparation, revision, and distribution of documents for specification test methods and release criteria, if not mentioned elsewhere (see also paragraph 1.9 and Chapter 4, Documentation).
Chapter 7. Contract Manufacture and Analysis
This chapter describes how GMP compliance of the contract acceptor is assessed, as well as the technical contract between the contract giver and acceptor, and the way in which the GMP compliance is assessed to ensure product compliance with the marketing authorization.
Chapter 8. Distribution, Complaints, and Product Recalls
• Arrangements and recording system for distribution (C.8.1): A description of the storage and distribution practices, ways of securing the warehouse, environmental controls of the warehouse, and how materials are stored is provided in this section. It is important to describe how the status of products is controlled (for example, by computer or by label), and how rejected materials are securely isolated. The authorities should be comfortable that the distribution records permit full batch traceability from the factory to the customer, in terms of the date of sale, customer details, and quantity dispatched, and that these records are readily available to permit an effective recall if required.
• Arrangements for the handling of complaints and product recalls (C.8.2): This section refers to the procedures for handling complaints and product recalls. Quality defects should be investigated and appropriate measures taken to prevent recurrence.
• Complaints (C.8.3): The person or persons responsible for logging, classifying, and investigating complaints is provided, along with information about written reports, their review and retention, and how causes of quality defects are investigated.
• Product recalls (C.8.4): Who is responsible for product recalls? Is there a written procedure that describes the sequence of actions to be followed (for example, retrieval of distribution data, notification to customers, receipt/segregation/inspection of returned product, investigation/reporting of cause, and reporting corrective action). Who notifies the authorities of complaints and recalls? Is the authority involved in complaints and the decision to recall? Can recalls be effective below wholesale level? List products recalled over the last two years. If none has been recalled, record “none.”
Chapter 9. Self-Inspection
• Include references to documented procedures that describe the self-inspection program and follow-up activities to self-inspections.
• Describe how the self-inspection system verifies that activities that have a bearing on quality comply with planned arrangements (that is, are records reviewed and activities observed to verify compliance with documents, procedures, and regulations?).
• State whether the program assesses the effectiveness of the quality system.
• Describe how the results of the self-inspection system are documented and brought to the attention of the personnel having responsibility for the area and activities inspected.
• Describe how the system ensures that those responsible for the area or activity take timely corrective action on the deficiencies found.
Drug Master File
A drug master file (DMF) is a submission to FDA of information, usually concerning the chemistry, manufacturing, and controls (CMC) of a component of a drug product, to permit FDA to review this information in support of a third party’s submission. A DMF may be used to provide confidential, detailed information about facilities, processes, or articles used in the manufacturing, processing, packaging, and storing of one or more human drugs. Drug product information or other non-CMC information may be filed in a DMF.
The submission of a DMF is not required by law or FDA regulation. A DMF is submitted solely at the discretion of the holder to maintain confidentiality of proprietary information (for example, manufacturing procedures) and to permit access to information by reviewers in the Center for Drug Evaluation and Research (CDER) to support applications submitted by one or more applicants. The information contained in the DMF may be used to support an Investigational New Drug Application (IND), a New Drug Application (NDA), an Abbreviated New Drug Application (ANDA), another DMF, an export application, or amendments and supplements to any of these.
A DMF is not a substitute for an IND, NDA, ANDA, or export application. It is not approved or disapproved. Technical contents of a DMF are reviewed only in connection with the review of an IND, NDA, ANDA, or export application. The DMF is reviewed only when it is referenced in an application or another DMF. The DMF is reviewed using the same regulatory and scientific criteria as review of an application.
Inspections of drug substance manufacturers usually are triggered when there is an application under review that references a DMF for the manufacture of that drug substance.
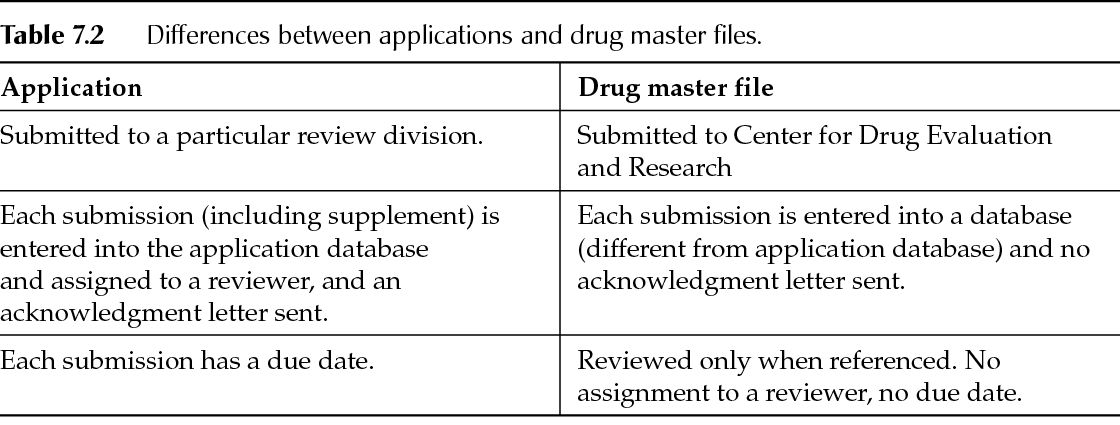
Confidentiality of information in DMFs is assured through 21 CFR 314.420(e). As DMFs usually cover manufacturing information, they usually are not considered releasable under a Freedom of Information Act (FOIA) request. Applications and DMFs are different (Table 7.2).
Types of Drug Master Files
Four types of DMFs have been identified (Table 7.3). More information on the number, type of DMF, and holder of the DMF are listed on the FDA website. The FDA website also provides detailed instructions and guidance for DMF submissions.

Changes to Drug Master Files
The DMF holder is expected to submit all changes to the DMF as amendments, notify FDA of change in holder name or address, notify FDA of change in agent/representative, submit an annual report or update, submit letter of authorization (LOA) for each item referenced for each customer, and notify authorized parties of changes. While annual updates to a DMF are not required by regulation, section VII of the DMF Guidelines recommends reporting a list of authorized parties, what they are authorized to reference, the date of the LOA, and a list of changes reported during the past year (that is, not a list of changes made, but a list of changes already reported). Stability updates should be reported as amendments.
If the anniversary date is missed, FDA will not send a reminder (unlike applications). If no changes have been made, it is important to send an update with a statement to that effect. It should be remembered that an amendment is a report of a change or addition of technical or administrative information. A supplement is not an amendment but only applies to approved applications. Pages that replace an already numbered page from a previous submission should also contain the page number in the current submission (for example, a page replacing page 10 in the original submission may be page 14 in the new submission). Finally, no pages are ever physically replaced in a DMF.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


