(1)
SUPA School of Physics and Astronomy, University of St. Andrews, St. Andrews, Scotland, UK
(2)
Faculté des Sciences, RNA Group, Département de Biologie, Université de Sherbrooke, Sherbrooke, QC, Canada
(3)
SUPA School of Physics and Astronomy, Biomedical Sciences Research Complex, University of St. Andrews, St. Andrews, Scotland, UK
Abstract
Single-molecule fluorescence studies of nucleic acids are revolutionizing our understanding of fundamental cellular processes related to DNA and RNA processing mechanisms. Detailed molecular insights into DNA repair, replication, transcription, and RNA folding and function are continuously being uncovered by using the full repertoire of single-molecule fluorescence techniques. The fundamental reason behind the stunning growth in the application of single-molecule techniques to study nucleic acid structure and dynamics is the unmatched ability of single-molecule fluorescence, and mostly single-molecule FRET, to resolve heterogeneous static and dynamic populations and identify transient and low-populated states without the need for sample synchronization. New advances in DNA and RNA synthesis, post-synthetic dye-labeling methods, immobilization and passivation strategies, improved dye photophysics, and standardized analysis methods have enabled the implementation of single-molecule techniques beyond specialized laboratories. In this chapter, we introduce the practical aspects of applying single-molecule techniques to investigate nucleic acid structure, dynamics, and function.
Key words
Single-molecule detectionDNARNAFörster resonance energy transfer (FRET)Total internal reflection (TIR)Fluorescence labeling1 Introduction
Over the past decade, single-molecule fluorescence spectroscopy has made new dynamic structural information about biological molecules available with an unprecedented level of detail. Single-molecule microscopy in combination with Förster resonance energy transfer (FRET) enables us to follow the conformational changes of spatially isolated nucleic acids, proteins, and protein-DNA or protein-RNA complexes over time periods ranging from milliseconds to minutes, thus covering the timescale of most biologically relevant processes. By isolating single biomolecules at low concentrations, transient or rare states that would be averaged over in a bulk experiment, altering the result or being lost, can be identified and quantified [1].
Single-molecule spectroscopy can be performed on low concentrations of sample diffusing freely in solution, termed fluorescence correlation spectroscopy (FCS), or immobilized on a substrate, generally a glass or quartz slide. Common to all single-molecule experiments is the requirement for a very small excitation volume to reduce background to a low enough level for fluorescence signals to be detectable. In FCS methods, excitation and detection take place at the focal spot of a laser, while with surface-immobilized systems, either a confocal point source or wide-field excitation using total internal reflection fluorescence microscopy (TIRF) can be used [1, 2]. The advantage of surface immobilization over freely diffusing techniques is that individual molecules can be observed for extended periods of time, limited only by the photostability of the reporter dyes, which has greatly improved with the introduction of efficient triplet-state quenchers and oxygen-scavenging systems [3, 4]. In this way, conformational dynamics for a variety of timescales and environments and entire folding pathways can be determined for each single molecule. This not only identifies transient states as mentioned above but also reveals the natural heterogeneity of folding pathways and lifetimes emerging in recent experiments involving complex nucleic acid structures [5, 6].
In this chapter, we will focus on the measurement of conformational dynamics in single surface-immobilized nucleic acids using FRET and wide-field TIRF techniques. We will describe the most widely used optical setups together with a detailed discussion of the protocols for post-synthetic site-specific labeling of nucleic acids with FRET pairs. Lastly, we will provide a comparative description of the software packages and analysis methods available to date to interpret single-molecule FRET trajectories.
1.1 Single-Molecule Förster Resonance Energy Transfer (smFRET)
Most single-molecule experiments on nucleic acid structure and function involve the measurement of conformational changes via FRET between two fluorophores covalently attached to known positions within the nucleic acid structure (see Note 1) (Fig. 1a), in which an optically excited donor (D) fluorophore transfers some of its energy to an acceptor (A) with an efficiency E app dependent on the interdye separation [7] (Fig. 1a, b). A trace of fluorescence intensity versus time for one molecule reveals not only structural changes but also their associated lifetimes, thus giving access to a wealth of dynamic information (Fig. 1c). Nucleic acids are particularly suited to surface-immobilized, extended-observation experiments because they do not interact as readily with the glass surface as proteins [8], which require additional surface passivation steps.
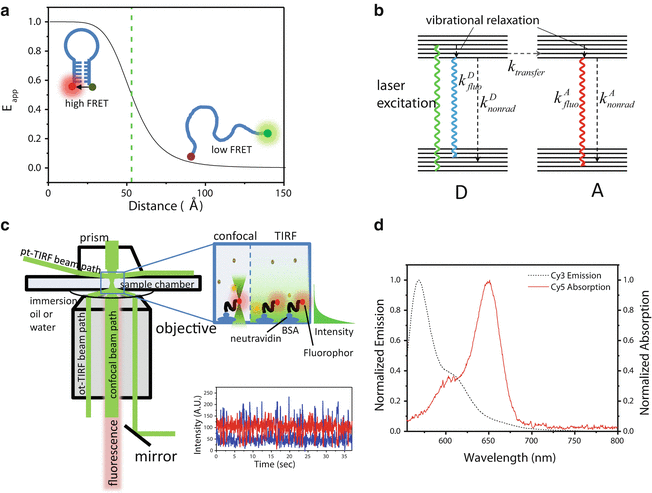
Fig. 1
Principle of FRET microscopy. (a) Efficiency of FRET as a function of interdye separation, R. E FRET depends on R −6, making changes in FRET-sensitive indicators of dynamics between molecules (intermolecular FRET) or between structural nucleic acid components tagged with fluorescent dyes (intramolecular FRET). (b) Jablonski diagram of energy transfer processes in FRET. On the left, a donor (D) molecule absorbs a photon and enters a higher electronic state. The molecule vibrationally relaxes to the lowest-energy sublevel, from which it de-excites either through fluorescence, nonradiative processes such as collisional, quenching, or through resonant energy transfer to the acceptor (A). Once excited, A can also decay through either radiative or nonradiative processes. (c) Schematic of the excitation and fluorescence pathways for TIR and single-molecule confocal microscopy. In pTIR, the excitation laser is focused onto the upper face of the sample chamber through a quartz prism (top left), while in oTIR, it is coupled onto the lower face through the extreme edge of the microscope objective (lower left). In confocal microscopy, the objective focuses the excitation beam as in ordinary confocal microscopy. In all cases, dye fluorescence is collected through the microscope objective, and residual excitation light is filtered or directed away from the detection optics. Right inset: excitation volumes for confocal and TIR microscopy. (Left) The confocal beam is scanned through the volume, illuminating one molecule at a time at the focal point. (Right) The evanescent wave illuminates a small area (~10 μm2) to a depth of about 150 nm. (d) Spectral overlap of Cy3 and Cy5 dyes, a common D–A pair. For the energy transfer process in (b) to be efficient, the dyes must be in close proximity (3–8 nm) and exhibit significant overlap between the emission spectrum of D and the absorption spectrum of A, as shown
The efficiency of FRET between a pair of fluorophores depends on the sixth power of the distance R between them, governed by the equation
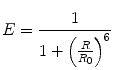
where R 0, the Förster radius, depends on the spectral properties and orientations of the dyes [7] (see Note 2). In practice, the apparent FRET efficiency, E app, is calculated from the ratio of acceptor fluorescence to total (donor + acceptor) fluorescence when only the donor is optically excited:
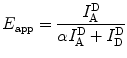
(1)
(2)
In Eq. 2, I A D is the intensity of acceptor fluorescence, I D D is the intensity of donor fluorescence, and α is a correction factor taking account of donor fluorescence leaking into the acceptor detection channel of the dyes. An additional correction factor accounting for differences in quantum yield and detection efficiency is needed to convert E app to the actual FRET efficiency E [7, 9]. The interdye separation can be calculated using Eq. 1, and relative differences in E app, E, or the related quantity ratio A (see Note 3), can be used to distinguish separation changes as small as 0.34 nm (1 base pair) [10]. The most widely used FRET pair for single-molecule studies contains a Cy3 dye as donor and a Cy5 as acceptor (Fig. 1d). Some reasons for the wide application of Cy3, Cy5, and their variants (i.e., Alexa, ATTO) in smFRET include their photostability (particularly in the presence of oxygen scavengers), appropriate spectral separation between donor and acceptor, similar quantum yield, and the fact that these dyes are commercially available with a variety of functional groups for coupling to both nucleic acids and proteins.
1.2 smFRET Studies of DNA and RNA Dynamics
In recent years, the application of smFRET to nucleic acids has revolutionized our understanding of DNA and RNA dynamics in vitro [11–14]. The variety of nucleic acid structures to which smFRET has been applied includes both single- and double-stranded RNA and DNA, DNA Holliday junctions [13, 15, 16] (Fig. 2a), guanine-quadruplexes (g-quadruplexes) [17–19] (Fig. 2b), and functional RNA motifs involved in catalysis (ribozymes; Fig. 2c–e) [11, 20–24] and gene regulation (riboswitches) (Fig. 2e–j) [25–32].
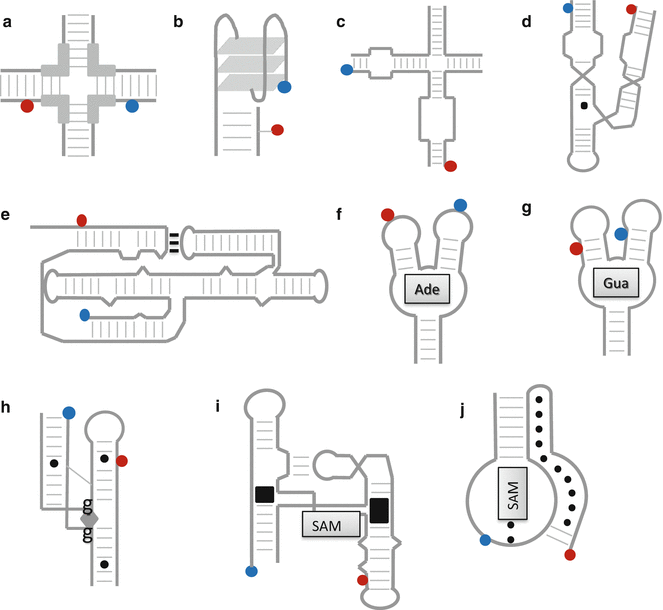
Fig. 2
Schematics of FRET experiments on nucleic acid structures. (a–j) illustrate the FRET configurations used to study the structural dynamics of several functional nucleic acid structures. (a) The mobile Holliday junction [13, 15, 16]. In this labeling configuration, E FRET changes when the branches migrate across the homologous region (thick lines), changing the separation between the dyes. (b) The guanine-quadruplex in the antiparallel strand configuration, which would yield a different FRET value from the parallel strand configuration, in which all four “corner” strands have the same directionality [19]. (c–e) Small catalytic ribozymes that play a central role in RNA processing by performing self-cleavage reactions. (c) The hairpin ribozyme [11, 23], (d) the hammerhead ribozyme [21, 24], and (e) the Varkud satellite (VS) ribozyme [22]. (f–j) Five sample riboswitches, 5′-untranslated mRNA sequences that regulate the expression of downstream genes via conformation changes induced by metabolite binding (metabolites pictured as shaded boxes). The riboswitches depicted here specifically bind (f) adenine [28, 32], (g) guanine [27], (h) c-di-GMP [31], (i) SAM-I [29], and (j) SAM-II [30]
A g-quadruplex is a three-tiered stack of guanosine tetramers that forms in regions of ssDNA (Fig. 2b) such as the telomere and the promoter regions of some DNAs [17]. Their appearance in these regions has given rise to proposals that they are involved in the inhibition of telomerase activity and in gene transcription, respectively [18]. smFRET has been used to follow the switching of telomeric g-quadruplexes between parallel and antiparallel strand configurations and to measure the lifetimes of the states at varying temperatures and ion concentrations [19]. Studies of promoter-region g-quadruplexes also confirmed that the g-quadruplex can form preferentially over the canonical double helix [17].
Another DNA structure that has received considerable interest because of its biological relevance is the four-way Holliday junction, a four-strand junction formed during DNA replication (Fig. 2a). smFRET studies have addressed both the conformational dynamics of its arms [13], which can stack in several different pairs, and its branch migration rate [15, 16]. Single-molecule confocal FRET was used to measure the distribution of step sizes along a homologous (migration-capable) sequence at one-base pair (bp) resolution and determine its sensitivity to nearby homologous sequences [16].
Three- and four-way helical junctions are also present in complex RNA structures like ribozymes, which are catalytically active RNA sequences that independently process RNA by self-cleavage and/or ligation [11, 20]. The Mg2+-induced folding pathway and catalytic activity of small ribozymes such as the hammerhead [21, 24], Varkud satellite (VS) [22], and hairpin ribozyme [11, 23] have been studied using single-molecule FRET. For instance, smFRET studies confirmed that the cleavage and ligation reactions of the hairpin ribozyme (Fig. 2c) require stable docking between loops on two of the four arms of a four-way junction [23].
Riboswitches are 5′-untranslated mRNA structures involved in gene regulation via ligand-induced structural changes (Fig. 2f–j). Binding of a ligand associated with the upstream gene triggers a structural change that either enhances or inhibits the formation of a promoter or terminator structure [25]. The contacts between the riboswitch and its ligand are often stabilized by tertiary interactions that enclose the ligand in a tight binding pocket [25, 26]. Surface-immobilized FRET experiments have addressed the rates of folding and unfolding in the presence of the ligand for a variety of structures including the riboswitches that sense guanine [27], adenine [28, 32], SAM-I [29], SAM-II [30], and cyclic GMP [31].
1.3 Single-Molecule Total Internal Reflection (sm-TIR)
The system under study and the information to be gained from it determine the most suitable single-molecule technique to use. In TIRF microscopy, a wide-field technique, the excitation light is coupled to the sample chamber at an angle greater than the critical angle for total internal reflection, resulting in an approx. 150 nm deep excitation volume illuminated by the evanescent wave (Fig. 1c) [33, 34]. The excitation light can be coupled either through a prism placed on top of the sample chamber or through the objective lens of the microscope, known as prism type and objective type (pTIR and oTIR), respectively. oTIR requires additional filtering of the excitation light from the fluorescence emission also collected by the objective, but high-numerical aperture (nA > 1.4) oil-immersion objectives reduce the noise cost formerly associated with oTIR [1]. oTIR also leaves the top of the chamber clear, making the simultaneous implementation of other techniques easier. In both forms of TIRF, all the molecules immobilized within a region approx. 10 μm2, usually 150–200 molecules, are simultaneously imaged on an EMCCD camera operating in the shot-noise limit at maximum frame rate 20–65 frames/s (15–50 ms time resolution) [33].
Scanning confocal microscopy raster-scans a focused excitation beam across the sample, exciting only one molecule at a time. It can scan continually, in which case time resolution is limited by the scanning speed, or stop at each molecule to record a time-dependent trajectory. Since all the detected photons are from the same molecule, a camera is not needed, and a fast APD detector can be used, increasing the time resolution into the sub-millisecond range [1]. In both TIRF and confocal microscopy, a longer integration time (longer time resolution) requires lower laser excitation power, which extends the lifetime of the fluorophores before photobleaching to several minutes or longer under continuous illumination [35].
1.4 Immobilization Methods for sm-TIR
The nucleic acid is usually chemically modified for surface immobilization by attaching a “tether” molecule called biotin (see Note 4). The sample chamber for TIRF is a microfluidic channel created between the coverslip and a microscope slide separated by two pieces of double-sided sticky tape. The chamber is successively coated with bovine serum albumin (BSA, a highly adherent protein complex) that has been labelled with biotin and then NeutrAvidin, which forms a very strong bond with biotin at one of its four binding sites (Fig. 3a). The biotin-labelled sample can then be flushed into the chamber and captured by the neutravidin [34]. The biotin-neutravidin interaction is resistant to moderate concentrations of denaturant, temperature variations, and illumination in the visible spectrum, making it ideal for most single-molecule fluorescence experiments. However, NeutrAvidin and derivatives are known to undergo chemical degradation under UV illumination.
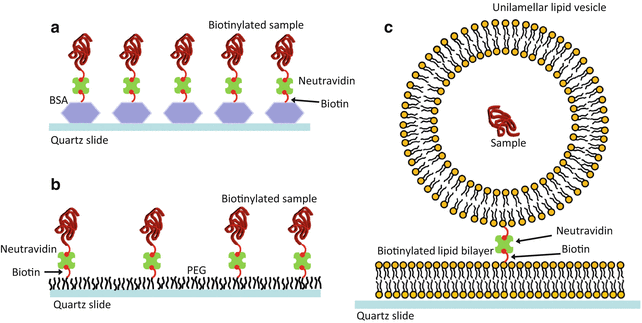
Fig. 3
Methods of sample immobilization. (a) Sample immobilization using the non-covalent interaction between biotin-labelled bovine serum albumin (BSA) and NeutrAvidin. The nucleic acid sample is also labelled with biotin, and pM concentrations are injected into the sample chamber and allowed to bind to another of the four NeutrAvidin binding sites. (b) To prevent nonspecific interactions between the slide and protein samples, the surface can be passivated with a layer composed of ~1 % biotinylated poly(ethylene glycol) (PEG) and non-biotinylated PEG before adding NeutrAvidin and the sample. (c) Samples that cannot be biotinylated can be encapsulated in porous lipid vesicles. The vesicles require a PEG layer or flat lipid bilayer (shown) to prevent them from bursting on contact with the quartz. A small fraction (<0.5 %) of the lipids are biotin labelled, and the “containers” are immobilized via the biotin-neutravidin interaction
If the nucleic acid structure does adhere to the glass surface or if the system under investigation involves protein-DNA interactions, it can be passivated using a combination of aminosilanization and polyethylene glycol (PEG) coating with a mixture of biotinylated and non-biotinylated PEG to prevent any nonspecific surface interactions (Fig. 3b). Finally, if tethering the sample molecule with biotin would severely limit its function or if it cannot accommodate enough modifications to include biotin, as is the case with many synthetic RNAs, each dye-labelled molecule can be enclosed in a biotin-labelled lipid vesicle approx. 100–200 nm in diameter, which is then tethered to biotin-neutravidin units on a pre-coated PEG slide or alternatively on a lipid bilayer passivated surface (Fig. 3c) [6, 36, 37].
1.5 Analysis of Time-Dependent Single-Molecule FRET Trajectories
Figure 4 shows typical single-molecule data in a variety of display formats. Figure 4a shows a raw intensity versus time (upper panel) and E app versus time (lower panel) trace for a single molecule, the output of the camera, or APD. (In the case of TIRF, each EMCCD movie must be analyzed by computer to identify the image of each molecule and read out the intensity record of those pixels.) The traces can be analyzed by eye or an automated routine and plotted to produce a histogram of population as a function of E app (Fig. 4b). A static population in a given physical state is expected to have a Gaussian E app distribution with the width determined by shot noise; broader or non-Gaussian peaks indicate the presence of dynamic interconversion between states on a timescale similar to the integration time [38].
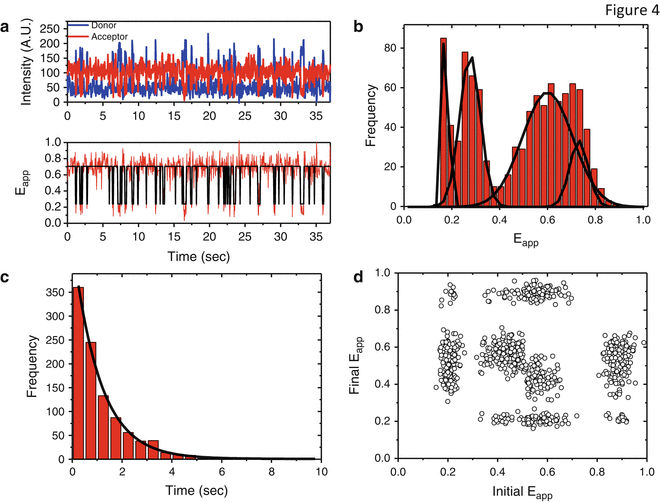
Fig. 4
Analysis of single-molecule data. (a, top panel) Representative single-molecule trace showing donor and acceptor variations in fluorescence intensity as a function of time for a nucleic acid sample exhibiting two-state dynamics. FRET efficiency can be calculated from the D and A intensities by Eq. 2, resulting in single-molecule trajectory of E app vs. time shown in the bottom panel. The black line indicates a hidden Markov fit performed with HaMMy, identifying the two FRET states between which the system fluctuates. (b) Representative single-molecule histogram of E app values drawn from a simulated four-state distribution. A histogram would be constructed from traces like (a) by taking the average FRET value of the first ten frames of each as the representative E app for that trace and constructing the histogram from them. A static, shot-noise-limited distribution should be accurately fit by a sum of Gaussian functions, so a non-Gaussian fit indicates dynamics faster than the time resolution of the experiment, leading to spurious FRET values, or a combination of very similar FRET states. (c) Lifetime measurement based on hidden Markov analysis. Following a fit by HaMMy or other Markov modeling software, the distribution of lifetimes of each state can be modeled and fitted by a single or multi-exponential decay curve e−t/τ, where τ is the lifetime of that state. (d) A transition density plot, showing all the initial and final states of transitions between the four states of the simulated data set in (b). A transition density plot can quickly display the frequency and direction of transitions between any pair of FRET states, and additional information, for example, about lifetime, can be included by color-coding the data points
Transition rates can be extracted from traces by fitting with a thresholding algorithm or, for complex systems with more than two or three states, hidden Markov modeling, as shown in Fig. 4a (lower panel). The dwell times of the states are then fit with a single or multi-exponential (Fig. 4c), whose time constant(s) corresponds to the rates of the transitions present in the system [39]. A two-dimensional population density plot (Fig. 4d) concisely represents the frequency of transitions between each pair of initial and final FRET states in the system [21, 39].
Surface-immobilized single-molecule techniques offer a valuable, detailed view of nucleic acid structural dynamics at a time when their importance as regulatory and catalytic structures within the cell is just beginning to be realized. In vitro techniques that can isolate important subpopulations and alternate folding pathways and probe environment- and sequence-dependent interactions are shedding light on the complex interactions that make the biological activity of ribozymes and riboswitches in physiological conditions possible. The development of high time resolution, high data throughput techniques is mitigating the perennial impact of dye photophysics on single-molecule experiments, increasing the resolution and reliability of the results. Finally, surface immobilization experimental techniques are easily interconvertible around a basic microscope and detection setup, allowing single-molecule labs great flexibility in the techniques they employ.
1.6 Data Analysis Software Packages
There is no specific commercial software available for single-molecule fluorescence analysis of TIR trajectories. Traditionally, most laboratories had to develop their own analysis software using MATLAB, Mathematica, or equivalent software packages. More recently, some of these packages have been made available to download and free to use by the single-molecule community. These programs use hidden Markov modeling (HMM) to generate and idealized trajectories based on the experimental one. A detailed review of the theory behind applying hidden Markov models to single-molecule data is given by Blanco and Walter [39].
2 Materials
2.1 Chemicals
2.1.1 Buffers
Several specific buffers are required for the labelling, purification, imaging, and long-term storage of nucleic acid structures. Here, we list the most commonly used buffers that can be obtained with high purity from most suppliers:
1.
Labelling buffer: coupling between a single strand of DNA or RNA carrying a primary amino group and a fluorescence dye containing a succinimidyl ester (NHS) is performed in 100 mM sodium tetraborate (Sigma) at pH 8.5 for optimal labelling. This buffer should be prepared as fresh as possible for maximum labelling efficiency.
2.
Ligation buffer: in those cases where the RNA or DNA motif is too long for chemical synthesis and it is not possible either to generate the DNA or RNA construct by hybridization of mixtures of single strands, ligation is the method of choice. The ligation of two strands by T4 DNA ligase or T4 RNA ligase requires a specialized buffer containing 5 mM ATP. Fermentas supplies this buffer with a purchase of T4 ligase, but it can also be prepared on site (see Subheading 3.3).
3.
Purification buffer: separation of labelled nucleic acids from free unreacted dye and unlabelled strands is normally performed via denaturing polyacrylamide gel electrophoresis (PAGE). For this 5× TBE is required containing 450 mM Tris-HCl, 450 mM borate, and 10 mM ethylenediaminetetraacetic acid disodium salt (EDTA).
4.
Pegylation buffer for surface functionalization: 10 mL ultrapure water (ddH2O) + 84 mg of sodium bicarbonate. This buffer should be freshly prepared every time to achieve maximum pegylation efficiency.
5.
Single–molecule imaging buffer: 50 mM Tris-HCl (pH 7.8), glucose in a (w/w) from 0.6 to 6 %, 1 % 2-mercaptoethanol, 0.1 mg/mL glucose oxidase type II-S from Aspergillus niger (Sigma), and 0.02 mg/mL glucose catalase (Sigma). 2-Mercaptoethanol can now be replaced by the more efficient triplet-state quencher Trolox (6-hydroxy-2,5,7,8-tetramethylchromane-2-carboxylic acid, Sigma).
2.1.2 Fluorophores
Small organic dyes are the most commonly used fluorophores for single-molecule experiments on nucleic acids. A good single-molecule fluorescent dye must be bright, photostable, and amenable to chemical modification for attachment to the nucleic acid (see Note 5). For FRET experiments, the D-A pair must also have a large (>100 nm) spectral separation between emission maxima, sufficient overlap between donor emission and acceptor excitation to promote efficient resonance energy transfer, and, when possible, similar quantum yields to produce unambiguous anticorrelated intensity changes [34]. Cy3 and Cy5 (GE Healthcare) and their corresponding Alexa (Invitrogen) or ATTO (ATTO-TEC) analogues are the most popular dyes for single-molecule studies (Tables 1 and 2 [40, 41]) because of their higher photostability and pH independence compared to fluorescein derivatives. They can be obtained with either succinimidyl modification (for coupling to a DNA or RNA carrying a primary amino group) or in maleimide format (for coupling to a DNA or RNA carrying a thiol modification).
Table 1
Spectroscopic properties and suppliers of fluorescence dyes suitable for single-molecule fluorescence
Dye | Abs. max (nm) | Em. max (nm) | ε at abs. max (M−1 cm−1) | Distributors | FRET partners |
|---|---|---|---|---|---|
Cy3 | 550 | 565 | 150,000 | GE, IDT | Cy5a, Cy5.5a, ATTO647Na, Fluoresceinb |
Cy5 | 655 | 667 | 250,000 | GE, IDT | Cy3b, Cy3bb, Cy5.5a |
Cy5.5 | 685 | 706 | 209,000 | GE, IDT | Cy3b, Cy5b |
AlexaFluor488 | 496 | 519 | 71,000 | Invitrogen | Alexa555a, Alexa647a |
AlexaFluor555 | 555 | 565 | 155,000 | Invitrogen | Alexa647 |
AlexaFluor647 | 650 | 670 | 239,000 | Invitrogen | Alexa488b, Alexa555b |
Fluorescein | 496 | 516 | 89,000 | IDT, Invitrogen | Cy3a |
TAMRA | 559 | 583 | 89,000 | IDT, Invitrogen | ATTO647Na, Cy5a |
Cy3b | 559 | 570 | 130,000 | GE | Cy5a, ATTO647Na |
ATTO647N | 644 | 669 | 150,000 | ATTO-TEC | TAMRAb, ATTO550b |
Table 2
R 0 for some single-molecule FRET pairs used in the cited literature
Donor | Acceptor | R 0 (nm) | Donor | Acceptor | R 0 (nm) |
|---|---|---|---|---|---|
Cy3 | Cy5 | 5.3 | Fluorescein | Cy3 | 5.6 |
Cy5 | Cy5.5 | 7.3 | TAMRA | Cy5 | 6.5 |
AlexaFluor555 | AlexaFluor647 | 5.1 | Cy3b | ATTO647N | 6.1 |
2.1.3 Dark Quenchers
Recently, non-emitting FRET acceptors known as Dark Quenchers (DQs) have been used instead of emissive acceptors to reduce noise in the data and free regions of the spectrum for additional dyes [42]. The properties of some Dark Quenchers appropriate for the most common FRET dyes are shown in Table 3 [43, 44]. Like fluorescent acceptors, DQs are prone to photoblinking and bleaching and need to be characterized accordingly before use [45]. Furthermore, to reliably distinguish quenching from donor normal intensity fluctuations (blinking, variations in quantum yield), triplet-state quenchers and oxygen-scavenging systems should be used to stabilize the donor emission (see Note 6), and the remaining baseline fluctuations should be well characterized. The primary advantage of DQs is that they permit an experiment to monitor three vectors simultaneously while using only two regions of the spectrum, simplifying the optical setup and data processing requirements. Also, they enable to investigate weak interactions which otherwise will be difficult to address by TIR due to the high amount of dye-labelled DNA/RNA or protein required.
Table 3
Spectroscopic properties for commonly used Dark Quenchers and corresponding suppliers
Quencher | Abs. max (nm) | ε (M−1 cm−1) | Effective quencher for dyes | Distributor |
|---|---|---|---|---|
BHQ1 | 534 | 34,000 | Fluorescein, rhodamine derivatives (TAMRA, TMR, Texas Red) | IDT |
BHQ2 | 578 | 38,000 | Cy and Alexa dyes | IDT |
QSY7 | 560 | 90,000 | Green-emitting dyes | Invitrogen |
QSY21 | 660 | 89,000 | Red-emitting dyes | Invitrogen |
2.1.4 Synthetic DNA or RNA
Companies such as Integrated DNA Technologies (IDT), Operon, Invitrogen, and Dharmacon (Thermo Scientific) offer custom DNA and RNA synthesis in nano- to micromolar scales of up to 140–150 nt length for DNA and slightly shorter for RNA. The exact 5′, 3′ and internal modifications that may be available will depend on the type of solid-phase synthesis (SPS) that each company provides. For RNA, additional deprotection steps may also be required depending on the company and the specific type of SPS chemistry used to manufacture the strands. For single-molecule immobilization studies, a biotin group is usually incorporated at the 5′ or 3′ end of one strand.
2.1.5 DNA Post-synthesis Fluorescent Labelling
Post-synthetic incorporation of fluorescent labels is normally performed using a succinimidyl ester (NHS) dye derivative and a synthetic nucleic acid strand carrying a primary amino group at a specific position. Although this is the most widely used approach and alternative, using a thiol modification and a maleimide derivative of the dye can also be performed. Incorporation of two specific dyes at known positions of a single strand is also possible using these two orthogonal chemistries. In this case, a single strand carrying a primary amino group and a thiol modification will be sequentially labelled with NHS- and maleimide-dye derivatives, respectively. For these labelling procedures, the following reagents are required:
1.
Synthetic single-stranded DNA or RNA carrying a primary amino and/or thiol modification (IDT, Operon, Thermo Scientific).
2.
Cy3 NHS or Cy3 maleimide or other modified dye carrying the appropriate coupling group.
3.
Dimethyl sulfoxide (e.g., Fisher Scientific).
5.
Precipitation solutions: 3 M sodium acetate and 100 % ethanol.
2.1.6 Purification of Fluorescently Labelled DNA or RNA by Polyacrylamide Gel Electrophoresis (PAGE)
2.
Dilution buffer 1×: 7 M urea dissolved in 1× TBE (store at room temperature).
3.
20 % acrylamide/bis solution (19:1) with 7 M urea in 1× TBE. Monomeric acrylamide is neurotoxic, and care should be taken to avoid exposure. Store this solution in the dark at room temperature.
4.
10 % (w/v) ammonium persulfate (APS): solution in water.
5.
TEMED (N,N,N′,N′-tetramethylethylenediamine).
6.
Running buffer (1× TBE).
7.
Molecular weight markers: xylene cyanol FF 0.02 % and bromophenol blue 0.02 % dissolved in pure formamide.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


