CHAPTER 14 Sideroblastic anemia
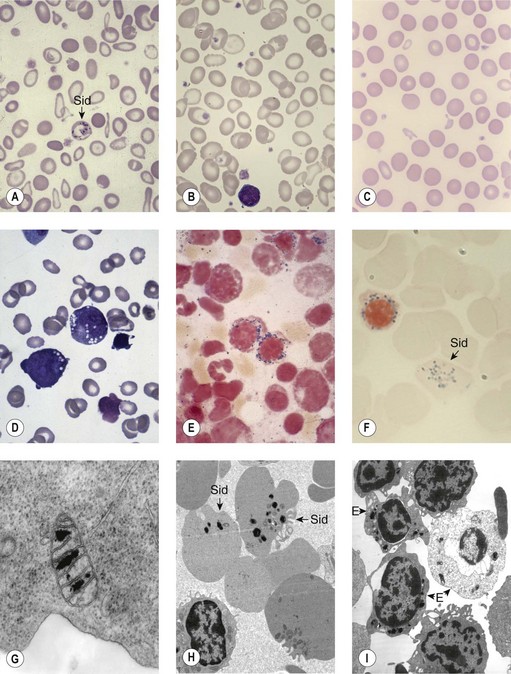
Sideroblastic anemias (SAs) are heterogeneous disorders characterized by accumulation of iron in the mitochondria of developing erythroblasts and varying degrees of red cell hypochromia. The abnormal, perinuclear mitochondria are visualized after Perls’ staining of bone marrow smears for iron. These are seen as a complete or partial ring of Prussian blue staining siderotic granules, in a substantial proportion of erythroblasts (Fig. 14.1).1,2 Recent studies further associate this excess iron with mitochondrial ferritin.3 SAs are inherited or acquired. The acquired forms may be secondary and reversible, or primary in nature.4
Hereditary sideroblastic anemia
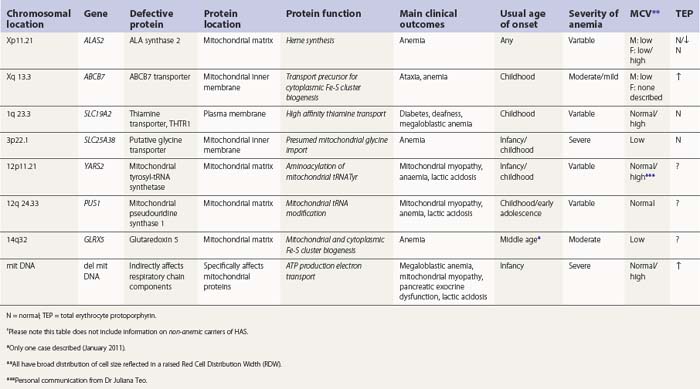
Hereditary SAs (HSAs) are very rare and all patterns of inheritance have been observed (Table 14.1). The anemia may be isolated or syndromic, and severity varies markedly. Some patients die in infancy or childhood; others have a normal life span. Although several genetic defects are now implicated a significant proportion of cases remain unexplained.5 The diagnosis is often achievable from clinical presentation and cell morphology but genetic diagnosis clarifies treatment options. This helps generate confident management plans which will avoid secondary complications and allows risk assessment for recurrence within families.
X-linked inheritance
ALA synthase 2 defect
The common form of X-linked SA (XLSA) is caused by mutations in the erythroid-specific, 5′-aminolevulinate synthase gene (ALAS2) at Xp11.21. Within the mitochondrial matrix, ALAS2 homodimer uses co-factor pyridoxal phosphate to catalyze the first step of heme synthesis.6 Decreased ALAS2 activity leads to decreased protoporphyrin production and decreased heme. Iron continues to enter the erythroblast unchecked, accumulates in the mitochondria and becomes most visible in the late erythroblasts on staining.
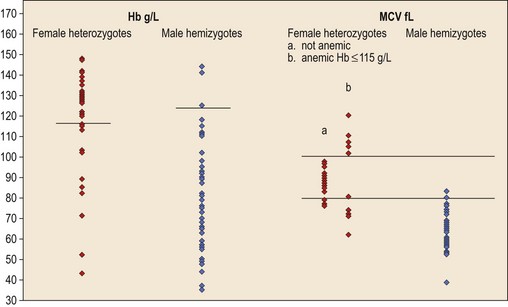
In male patients the severity of the anemia is variable. It is microcytic-hypochromic and may respond to pyridoxine treatment (Fig. 14.1A, B).7,8 Female carriers have the potential to produce two RBC populations, one microcytic-hypochromic and one normal, depending on which erythroblast X-chromosome is active (Fig. 14.1C). Carriers are usually unaffected but some are anemic due to skewed X chromosome inactivation against that carrying the normal allele, accounting for almost one third of probands.9,10 Anemic carriers of mild or moderately severe mutations have the same presentation as male hemizygotes (Fig. 14.2). Patients can present at any age with symptoms of anemia or iron overload.4,11
Physical examination is unremarkable although mild hepatosplenomegaly may be present. Red cell size and hemoglobin content are broadly distributed and variable proportions of microcytic-hypochromic and normocytic-normochromic cells are seen on the blood film (Fig. 14.1A,B). Anisocytosis, poikilocytosis, elliptocytosis and target cells may be present. An occasional cell with basophilic stippling or Pappenheimer bodies and an occasional late erythroblast may be seen; the numbers of these increase after splenectomy. White cell and platelet counts are normal.
In the bone marrow erythropoiesis is expanded but ineffective; iron overload from increased dietary iron absorption may develop. Transferrin saturation is usually increased; even mildly anemic patients may present with secondary iron overload in the absence of blood transfusion.12 Total erythrocyte protoporphyrin (TEP) is low/normal. Severe anemia requiring regular blood transfusions is a rare occurrence. Phlebotomy has been successful for preventing iron overload, reversing iron overload and maintaining normal iron levels in pyridoxine-responsive patients (see below). This may additionally improve the hemoglobin level.13 Iron chelation may be required for correcting iron overload. Splenectomy is not recommended because this has led to thrombotic complications.4
At least 120 cases from 70–80 families are reported (January 2011). More than 50 different mutations are involved, scattered across seven exons encoding the catalytic and C-terminal domains.4,5 Missense mutations predominate; null mutations, or those predicted to be very severe, are found only in female heterozygotes. ALAS2 variations causing anemia in one member of a family may occasionally be silent in another.14
Pyridoxal phosphate (vitamin B6) is required for ALAS2 activity and stability. Many patients respond partially to pharmacologic doses of oral pyridoxine but some do not or barely do so.13 Responsive patients remain on maintenance doses of about 25–200 mg oral pyridoxine/day for life.4 Complete correction of hematological changes with pyridoxine, although rare, has been reported.15 Pyridoxine responsiveness is most associated with mutations involving amino acids fairly close to the pyridoxal phosphate binding site that cause partial loss of enzyme function or stability.16,17 Refractoriness is associated with mutations that cause irreversible disruption of activity or cause loss of in vivo activity through faulty interaction with components required for intramitochondrial processing.18 Iron overload, or its complications, also contribute to pyridoxine refractoriness. There may be some response to folate due to secondary deficiency.
Not all ALAS2 mutations cause SA. Mutations leading to deletion or substitution of the C-terminal 19 amino acids generate truncated ALAS2 protein of increased activity and cause X-linked, dominant erythropoietic protoporphyria, not anemia.19
Female carriers of severe/null mutations that prevent erythrocyte production go undetected unless they become anemic because of either skewed but incomplete X-chromosome inactivation against the normal allele or some mechanism selecting against cells expressing the normal allele. In these cases red cells produced by residual effective erythropoiesis from expression of normal ALAS2 are macrocytic, the number of ring sideroblasts may be very low and the cause of the ineffective erythropoiesis may go unsuspected or misdiagnosed (Fig. 14.2).10,20
ABCB7 defect
A second XLSA is associated with slow/non-progressive, cerebellar ataxia (XLSA/A). This is usually diagnosed in infants and young children who present with delayed walking ability.21 There is mild to moderate microcytic-hypochromic anemia (Hb 8–15 g/dl) with red cell appearances indistinguishable to those of ALAS2 deficiency. However, in contrast, there are features of iron deficiency (normal/decreased serum iron, normal/low transferrin iron saturation, increased serum transferrin receptor concentration and variably increased TEP predominantly of the zinc form) rather than iron overload.22–24 Elevated serum ferritin did, however, develop in two patients, despite low/normal transferrin saturations, so monitoring iron levels is warranted.24 Ferrokinetic studies have not been reported and bone marrow reports do not give a consistent picture. Down-regulation of ABCB7 in non-erythroid cells decreases cell proliferation;25 erythroid hypoplasia may play a role therefore in the anemia. Rational management and treatment of the unusual distribution of iron in these patients awaits a better understanding of the molecular aetiology.
Brain magnetic resonance imaging (MRI) shows selective cerebellar atrophy (Fig. 14.3A). Dysmetria, finger-nose and heel-shin ataxia, dysarthria, intention tremor and diminished deep tendon reflexes are usually present to varying degrees. Nystagmus, strabismus and abnormal plantar responses may or may not be present. Early difficulty in sitting and walking is often followed by some improvement over time. Intelligence is normal and there is no sensory loss.21,26
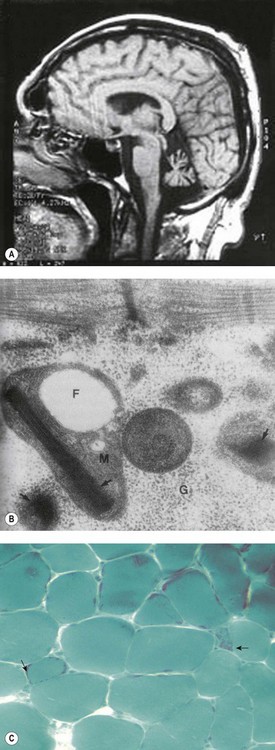
Fig. 14.3 (A–C) Syndromic sideroblastic anaemia: (A) MRI scan of the brain of a patient with XLSA/A showing cerebellar atrophy.26 (B, C) Mitochondrial inclusions and ragged red fibers in a muscle biopsy of a patient with myopathy with lactic acidosis and sideroblastic anemia.32,34
(These figures are reproduced by permission of (A) BMJ Publishing Group Ltd @ 2001, license number 2382030149869, (B) John Wiley and Sons Inc. @ 2005, license number 2382570556744 and (C) BMJ Publishing Group Ltd @ 2007, license number 2382021149604.)
The defect lies in ABCB7 at Xq13.3 encoding an ATP-dependent ‘half-transporter’ consisting of N-terminal transmembrane transporter and C-terminal ATPase domains. Within the inner mitochondrial membrane, ABCB7 homodimer exports some component essential for cytoplasmic Fe-S cluster formation.27 Four separate families with 12 affected patients have been reported. In each of the three families studied a different missense mutation was detected (I400M, V411L, E433K) affecting a different amino acid within the transmembrane transport domain. Yeast cells lacking homologous Atm1p, show reduced cytoplasmic Fe-S cluster generation, normal mitochondrion Fe-S, and mitochondrial iron overload. This phenotype was partially reversed by the intracellular expression of human ABCB7 but less so by the E433K mutant, implicating loss of cytoplasmic Fe-S cluster generation in the XLSA/A syndrome.23
Cellular iron homeostasis in humans is regulated at the translational level through the interaction of two iron regulatory proteins (IRP1 and IRP2) with mRNA hairpin-like secondary structure elements known as iron response elements (IRE). IRP1 is formed by loss of Fe-S from cytoplasmic (c-) aconitase. ALAS2 mRNA has a 5′ IRE and protein levels are controlled at the translation stage by iron.28 Low cytoplasm Fe-S decreases ALAS2 activity through conversion of c-aconitase (4Fe-4S) to IRP1 that binds to the 5′ IRE and blocks translation. This explains microcytic-hypochromic anemia and mitochondrial iron loading with ABCB7 deficiency; however, additionally, excess mitochondrial iron is somehow not available to ferrochelatase that uses zinc instead to form zinc-protoporphyrin.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


