Sickle Cell Disease
KEY CONCEPTS
![]() Sickle cell disease (SCD) is an inherited disorder caused by a defect in the gene for β-globin, a component of hemoglobin, and is called a qualitative hemoglobinopathy. Patients can have one defective gene (sickle cell trait that is not a disease) or two defective genes (SCD).
Sickle cell disease (SCD) is an inherited disorder caused by a defect in the gene for β-globin, a component of hemoglobin, and is called a qualitative hemoglobinopathy. Patients can have one defective gene (sickle cell trait that is not a disease) or two defective genes (SCD).
![]() Although SCD usually occurs in persons of African ancestry, other ethnic groups can be affected. Multiple mutation variants result in different clinical manifestations.
Although SCD usually occurs in persons of African ancestry, other ethnic groups can be affected. Multiple mutation variants result in different clinical manifestations.
![]() SCD involves multiple organ systems. Usual clinical signs and symptoms include anemia, pain, splenomegaly, and pulmonary symptoms. SCD can be identified by routine neonatal screening programs. Early diagnosis allows early comprehensive care.
SCD involves multiple organ systems. Usual clinical signs and symptoms include anemia, pain, splenomegaly, and pulmonary symptoms. SCD can be identified by routine neonatal screening programs. Early diagnosis allows early comprehensive care.
![]() Patients with SCD are at risk for infection. Prophylaxis against pneumococcal infection reduces death during childhood in children with sickle cell anemia.
Patients with SCD are at risk for infection. Prophylaxis against pneumococcal infection reduces death during childhood in children with sickle cell anemia.
![]() Hydroxyurea decreases the incidence of painful episodes, but patients treated with hydroxyurea should be carefully monitored.
Hydroxyurea decreases the incidence of painful episodes, but patients treated with hydroxyurea should be carefully monitored.
![]() Neurologic complications caused by vasoocclusion can lead to stroke. Screening with transcranial Doppler ultrasound to identify children at risk accompanied by chronic transfusion therapy programs have been shown to be beneficial in decreasing the occurrence of overt stroke in children with SCD.
Neurologic complications caused by vasoocclusion can lead to stroke. Screening with transcranial Doppler ultrasound to identify children at risk accompanied by chronic transfusion therapy programs have been shown to be beneficial in decreasing the occurrence of overt stroke in children with SCD.
![]() Patients with fever greater than 38.5°C (101.3°F) should be evaluated, and appropriate antibiotics administered immediately, including coverage for encapsulated organisms, especially pneumococcal organisms.
Patients with fever greater than 38.5°C (101.3°F) should be evaluated, and appropriate antibiotics administered immediately, including coverage for encapsulated organisms, especially pneumococcal organisms.
![]() Pain episodes can often be managed at home. Hospitalized patients require parenteral analgesics. Analgesic options include opioids, nonsteroidal antiinflammatory agents, and acetaminophen. The patient characteristics and the severity of the pain should determine the choice of agent and regimen.
Pain episodes can often be managed at home. Hospitalized patients require parenteral analgesics. Analgesic options include opioids, nonsteroidal antiinflammatory agents, and acetaminophen. The patient characteristics and the severity of the pain should determine the choice of agent and regimen.
![]() Patients with SCD should be followed regularly for healthcare maintenance issues and monitored for changes in organ functions.
Patients with SCD should be followed regularly for healthcare maintenance issues and monitored for changes in organ functions.
![]() Sickle cell syndromes, which can be divided into sickle cell trait (SCT) and sickle cell disease (SCD), are a group of hereditary conditions characterized by the presence of sickle cell hemoglobin (HbS) in red blood cells (RBCs). SCT is the heterozygous inheritance of one normal β-globin gene producing HbA and one sickle gene producing HbS (HbAS) gene. Individuals with SCT are asymptomatic. SCD can be of homozygous or compounded heterozygous inheritance. Homozygous sickle cell hemoglobin (hemoglobin S) (HbSS) has historically been referred to as sickle cell anemia (SCA). The heterozygous inheritance of HbS with another qualitative or quantitative β-globin mutation results in sickle cell hemoglobin C (HbSC), sickle cell β-thalassemia (HbSβ+-thal and HbSβ0-thal), and some other rare phenotypes.1,2
Sickle cell syndromes, which can be divided into sickle cell trait (SCT) and sickle cell disease (SCD), are a group of hereditary conditions characterized by the presence of sickle cell hemoglobin (HbS) in red blood cells (RBCs). SCT is the heterozygous inheritance of one normal β-globin gene producing HbA and one sickle gene producing HbS (HbAS) gene. Individuals with SCT are asymptomatic. SCD can be of homozygous or compounded heterozygous inheritance. Homozygous sickle cell hemoglobin (hemoglobin S) (HbSS) has historically been referred to as sickle cell anemia (SCA). The heterozygous inheritance of HbS with another qualitative or quantitative β-globin mutation results in sickle cell hemoglobin C (HbSC), sickle cell β-thalassemia (HbSβ+-thal and HbSβ0-thal), and some other rare phenotypes.1,2
Over the years, progress has been made in understanding the relationship between clinical severity and genotype, as well as the natural history of common morbidities associated with SCD. Ongoing research involves investigation of pharmacotherapies used to treat SCD. Recent advances in the care of SCD patients have increased life expectancy.1–7
SCD is a chronic illness with significant burden for family and society. Frequent hospitalizations can interrupt schooling and result in employment difficulties.8–10 Acute complications of the disease can be unpredictable, rapidly progressive, and life threatening. Later in life, chronic organ damage and cognitive or emotional impairments can develop.1,2 Because of the complexity and gravity of the illness, it is essential that comprehensive care is available to all patients and that all providers involved have a good understanding of the disease and its management.1,2,8,11
EPIDEMIOLOGY
![]() SCD affects millions of people worldwide and is most common in people with African heritage.1,12 The most common SCD genotype is HbSS (~60% to 65%), followed by HbSC (~25% to 30%), HbSβ+-thal and HbSβ0-thal (~5% to 10%). Other variants account for less than 1% of patients.1,2 The disease is common among those with ancestors from sub-Saharan Africa, India, Saudi Arabia, and Mediterranean countries.2,13 In the United States, about 90,000 Americans have SCD with a prevalence of 1 in 2,500 newborns, 1 in 500 African Americans, and 1 in 36,000 Hispanic births.1,14,15 About 2 million Americans have SCT with a prevalence rate of 1 in 12 African Americans and 1 in 100 Hispanics.16
SCD affects millions of people worldwide and is most common in people with African heritage.1,12 The most common SCD genotype is HbSS (~60% to 65%), followed by HbSC (~25% to 30%), HbSβ+-thal and HbSβ0-thal (~5% to 10%). Other variants account for less than 1% of patients.1,2 The disease is common among those with ancestors from sub-Saharan Africa, India, Saudi Arabia, and Mediterranean countries.2,13 In the United States, about 90,000 Americans have SCD with a prevalence of 1 in 2,500 newborns, 1 in 500 African Americans, and 1 in 36,000 Hispanic births.1,14,15 About 2 million Americans have SCT with a prevalence rate of 1 in 12 African Americans and 1 in 100 Hispanics.16
About 275,000 babies are born with SCD every year and 85% occur in Africa.2,12 The prevalence of SCD in the region is determined by the frequencies of SCT. The distribution of SCT reflects the survival advantage in regions (tropical areas) where malaria is endemic as the gene mutation offers partial protection against serious malarial infection. RBCs carrying the abnormal sickle hemoglobin prevent the normal growth and development of Plasmodium falciparum within RBCs. Individuals with SCT are more likely to survive the acute malarial illness whereas individuals with SCD-HbSS often present with more severe disease. The incidence of the sickle gene in a population correlates with the historical incidence of malaria and SCT results in partial resistance to the disease.1,2,12,17
The prevalence of SCD is highest in sub-Saharan Africa. Other areas where the sickle mutation can be found include the Arabian Peninsula, the Indian subcontinent, and the Mediterranean region. In Africa, the variants are Senegal (Atlantic West Africa), Benin (Central West Africa), Bantu (Central African Republic), and Cameroon. Arab-Indian haplotype is seen in certain areas of Saudi Arabia and India.2,13 Haplotypes identified through newborn screening programs in the United States showed that the Benin haplotype was the most frequent (63%), followed by Bantu (14%), Senegal (9%), Cameroon (4%), and Saudi Arabian (2%).13 Genetic analysis shows that the mutation found in Arabic patients is different from the mutation in those of African descent. Sickle gene mutation variants have been associated with different geographic locations and may be responsible for variations in clinical manifestations.3,5,13
ETIOLOGY
Normal hemoglobin (hemoglobin A [HbA]) is composed of two α chains and two β chains (α2 β2). The biochemical defect that leads to the development of HbS involves the substitution of valine for glutamic acid as the sixth amino acid in the β-polypeptide chain. Another type of abnormal hemoglobin, hemoglobin C (HbC), is produced by the substitution of lysine for glutamic acid as the sixth amino acid in the β-chain. Structurally, the α-chains of HbS, HbA, and HbC are identical. Therefore, it is the chemical differences in the β-chain that account for sickling and its related sequelae.1–3
SCA or HbSS is the most common form of SCD and occurs when an individual inherits both maternal and paternal β-globin alleles that code for the HbS Figures 82-1 to 82-4 show the probability of inheritance with each pregnancy for the offspring of parents with HbA, SCT, and HbSS. If both parents are carriers, the offspring will have a 25% risk of inheriting SCD and a 50% risk of SCT (Fig. 82–1). β-Thalassemia is a quantitative hemoglobinopathy resulting from a genetic defect in β-globin production. β-Thalassemia can be co-inherited with HbS and may vary from no β-globin production (β0) to some β-globin production (β+). Individuals with HbSS and HbSβ0-thal have a more severe course than those with HbSC and HbSβ+-thal.2,13 As discussed earlier, several haplotypes characterize the sickle gene, resulting in different clinical and hematologic courses. Included among these types are the three most commonly found in the United States: the Bantu haplotype, characterized by severe disease; the Senegal haplotype, characterized by mild disease; and the Benin haplotype, characterized by a course intermediate to that of the other two haplotypes. Although there are a number of other haplotypes seen around the world, the major types outside of the United States include Saudi Arabian and Cameroon, both with milder courses of illness.2,3,5
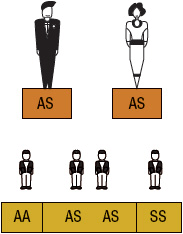
FIGURE 82-1 Sickle cell gene inheritance scheme for both parents with sickle cell trait (SCT). Possibilities with each pregnancy: 25% normal (AA); 50% SCT (AS); and 25% sickle cell anemia (SS). (A, normal hemoglobin; S, sickle cell hemoglobin)
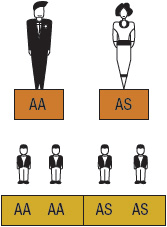
FIGURE 82-2 Sickle cell gene inheritance scheme for one parent with sickle cell trait (SCT) and one parent with no sickle cell gene. Possibilities with each pregnancy: 50% normal (AA); 50% SCT (AS). (A, normal hemoglobin; S, sickle cell hemoglobin)
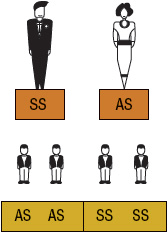
FIGURE 82-3 Sickle cell gene inheritance scheme for one parent with sickle cell trait (SCT) and one parent with sickle cell anemia (SCA). Possibilities with each pregnancy: 50% SCA (SS); 50% SCT (AS). (A, normal hemoglobin; S, sickle cell hemoglobin)
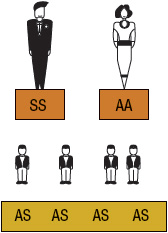
FIGURE 82-4 Sickle cell inheritance scheme for one parent without sickle cell gene and one parent with sickle cell anemia (SCA). Possibilities with each pregnancy: 100% SCT (AS). (A, normal hemoglobin; S, sickle cell hemoglobin)
PATHOPHYSIOLOGY
Normal adult RBCs contain predominantly HbA (96% to 98%). Other forms of hemoglobin are HbA2 (2% to 3%) and fetal hemoglobin (HbF; less than 1%). Normal RBCs are biconcave shape and able to deform to squeeze through capillaries.1–3,18 HbF is present predominantly in fetal RBCs and is a tetramer of two α globin chains and two γ globin chains (α2γ2). Prior to birth, HbF is the predominant hemoglobin type. Around 32 weeks gestation, a switch from the production of γ chains to β chains occurs and, consequently, an increase in HbA production is seen. Increased HbF production is seen under severe erythroid stress, such as anemia, hematopoietic stem cell transplantation (HSCT), or chemotherapy or in the hereditary condition, hereditary persistence of fetal hemoglobin (HPFH) where a mutation in the β-globin gene cluster results in continued HbF production after birth. HPFH is a benign, asymptomatic condition.3,5,7,19
In the pathogenesis of SCD, the following are primarily responsible for the various clinical manifestations: impaired circulation, destruction of RBCs, and stasis of blood flow. These changes result directly from two major disturbances involving RBCs: abnormal hemoglobin polymerization and membrane damage Fig. 82–5).
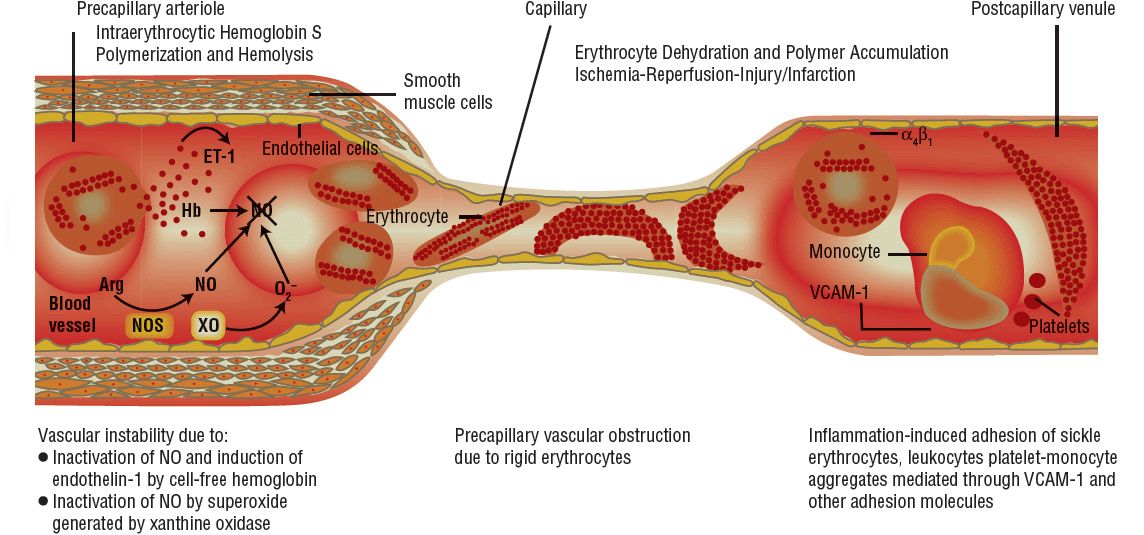
FIGURE 82-5 Pathophysiology of sickle cell disease. (Arg, arginine; ET-1, endothelin-1; Hb, hemoglobin; NO, nitric oxide; NOS, nitrous oxide synthase; VCAM-1, vascular cell adhesion molecule 1; XO, xanthine oxidase.) (From Kato GJ, Gladwin MT. Sickle cell disease. In: Hall JB, Schmidt GA, Wood LDH, eds. Principles of Critical Care, 3rd ed. New York: McGraw-Hill, 2005:1658.)
The solubilities of HbS and HbA are the same under conditions of normal oxygenation. Because of increased hydrophobicity as a result of the valine-substituting glutamic acid substitution, solubility of deoxygenated HbS is reduced. Saturation of deoxy-HbS leads to intermolecular binding and formation of thin bundles of fibers, which initially are unstable. However, the increased binding of deoxy-HbS eventually results in cross-linked fibers and stable polymers. This process is influenced by mean corpuscular hemoglobin concentration (MCHC), temperature, intracellular pH, and the circulating amount of HbS. Polymerization allows deoxygenated hemoglobin molecules to exist as a semisolid gel that protrudes into the cell membrane, leading to distortion of RBCs (sickle shaped) and loss of deformability. The presence of sickled RBCs increases blood viscosity and encourages sludging in the capillaries and postcapillary venules. Such obstructive events lead to local tissue hypoxia, which tends to accentuate the pathologic process.1,2,6
When reoxygenated, polymers within the RBCs are lost, the RBCs eventually return to normal shape. This process contributes to the vasoocclusive manifestation in that HbS is able to squeeze into microvasculature when oxygenated, but becomes sickled when deoxygenated. The cycle of sickling and unsickling results in damage to the cell membrane, loss of membrane flexibility, and rearrangement of surface phospholipids. Membrane damage also alters ion transport, resulting in potassium and water loss, which can lead to a dehydrated state that enhancing the formation of sickled forms. After continual repetitions of the process, the RBC membrane develops into rigid irreversibly sickled cell (ISC). Unlike the reversible sickled cells, which have normal morphology when oxygenated, ISCs are elongated cells and remain sickled when oxygenated. More rigid membranes of HbS-containing RBCs retard flow, particularly through the microcirculation. In addition, sickled RBCs tend to adhere to vascular endothelial cells, which further increase polymerization and obstruction.1,2,6
Intermolecular binding and polymer formation are reduced by HbF and to a lesser degree by HbA2. RBCs that contain HbF sickle less readily than cells without. ISCs, not surprisingly, have a low HbF level. Increased levels of HbF, as in the case of the Saudi Arabian genotype, result in more benign forms of SCD. The amount of HbF and HbA2 in relation to HbS influences the clinical manifestations and accounts for some of the variability in severity among SCD genotypes.2,5
Intravascular destruction of sickle cells can occur at an accelerated rate. The stresses of circulation and repetitive sickle–unsickle cycles lead to cell fragmentation. Damage to the cell membrane promotes cell recognition by macrophages. Rigid ISCs are easily trapped, resulting in short circulatory survival and chronic hemolysis. The typical sickled cell survives for about 16 to 20 days, whereas the life span of a normal RBC is 120 days. Anemia triggers the release of immature RBCs (reticuloctyes) from bone marrow prematurely. Surface adhesion proteins that maintain the reticulocytes inside the marrow adhere to the endothelium in postcapillary venules, further blocking the mature HbS-containing RBCs leading to complete occlusion of microvessels.6,18
In addition to sickling, other factors are also responsible for the clinical manifestations associated with SCD. Hemolysis releases free hemoglobin resulting in generation of reactive oxygen species, and alternation of nitric oxide (NO) metabolism. Obstruction of blood flow to the spleen by sickle cells can result in functional asplenia, defined as the loss of splenic function with an intact spleen. These patients can also have deficient opsonization. Impaired splenic function increases susceptibility to infection by encapsulated organisms, particularly pneumococcal bacteria. Coagulation abnormalities in SCD can be the result of continuous activation of the hemostatic system or disorganization of the membrane layer.2,6
CLINICAL PRESENTATION
![]() SCD is usually identified by routine neonatal screening programs in the United States. Since 2006, universal neonatal screening for SCD is performed in all 50 states. The sensitivity and specificity of screening methods (isoelectric focusing high-performance liquid chromatography, or electrophoresis) approach 100%.20 For infants with a positive screening result, a second test should be performed by 2 months of age to confirm the diagnosis. More than 98% newborns in the United States are screened for SCD to identify the disease. Despite universal screening, some infants with SCD are not identified at birth because of extreme prematurity, prior blood transfusion, or inability to contact family.19–21
SCD is usually identified by routine neonatal screening programs in the United States. Since 2006, universal neonatal screening for SCD is performed in all 50 states. The sensitivity and specificity of screening methods (isoelectric focusing high-performance liquid chromatography, or electrophoresis) approach 100%.20 For infants with a positive screening result, a second test should be performed by 2 months of age to confirm the diagnosis. More than 98% newborns in the United States are screened for SCD to identify the disease. Despite universal screening, some infants with SCD are not identified at birth because of extreme prematurity, prior blood transfusion, or inability to contact family.19–21
SCD involves multiple organ systems, and its clinical manifestations vary greatly between genotypes Table 82–1). Persons with SCT are usually asymptomatic and SCT is not considered a disease. However, under certain extreme situations where hemoglobin oxygenation is altered, RBC sickling can occur. Individuals with SCT should be cautious when participating in exercise under extreme conditions, such as high altitude or military training. Sickling of RBCs in the renal medulla, an area with low oxygen tension, can result in the inability to concentrate urine. Individuals with such impairment are at risk of dehydration during periods in which the body normally conserves water. Microscopic hematuria has been observed, and gross hematuria can occur after heavy exercise. Other reported complications associated with SCT are venous thromboembolism, renal medullary carcinoma, and chronic kidney disease.14,17,22 An increased incidence of urinary tract infection in women, especially during pregnancy, was previously reported and specific screening guidelines should be followed.23
TABLE 82-1 Clinical Features of Sickle Cell Trait and Common Types of Sickle Cell Disease
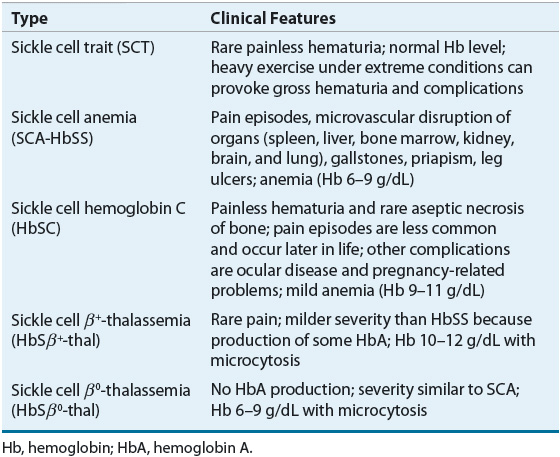
The cardinal features of SCD are hemolytic anemia and vasoocclusion. In individuals with HbSS, anemia usually develops from 4 to 6 months after birth. The delay in presentation is due to the presence of HbF in the infant’s RBCs. However, HbF production is gradually replaced by HbS, which typically leads the clinical manifestations of hemolysis and vasoocclusion, such as pain and swelling of the hands and feet, commonly referred to as hand-and-foot syndrome or dactylitis in infants.1,2
The common clinical signs and symptoms associated with HbSS include chronic anemia and pallor; fever; arthralgia; scleral icterus; abdominal pain; weakness; anorexia; fatigue; enlargement of the liver, spleen, and heart; and hematuria. Laboratory findings include the low hemoglobin level around 6 to 9 g/dL (3.72 to 5.58 mmol/L), elevated reticulocytes of 10% to 20%, and elevated platelet and white blood cell (WBC) counts. Mean corpuscular volume (MCV) is normal. The peripheral blood smear demonstrates sickled red cell forms.1,14,18
Individuals with HbSC disease present with less severe symptoms than that of HbSS and can be characterized primarily by mild anemia (hemoglobin levels of 9 to 11 g/dL [5.58 to 6.82 mmol/L] and reticulocytes of 3% to 10%), infrequent episodes of pain, persistence of splenomegaly into adult life, and excessive target cells in the peripheral blood smear. In individuals with heterozygous HbS-β-thalassemia syndrome, severity of disease depends on the thalassemia gene involved.1,18
The Cooperative Study of Sickle Cell Disease has previously reported that predictors for severe disease in children are dactylitis before 1 year of age, an average hemoglobin less than 7 g/dL (4.34 mmol/L) in the second year of life, and leukocytosis in the absence of infection. However, these variables could not be validated in a more recent study.24 Early acute chest syndrome (ACS) during the first 3 years of life is a predictor for recurrent episodes throughout childhood. Children with concomitant SCD and asthma have increased frequencies of ACS and pain episodes and increased mortality. Risk factors for early death in adults with SCD include complications such as sickle cell pain, anemic events, ACS, renal failure, and pulmonary disease.1,25–27 With longer survival for SCD, chronic manifestations of the disease contribute to the increased prevalence of morbidity later in life.
COMPLICATIONS
Acute Complications
Fever and Infection
Functional asplenia and failure to make antibodies against encapsulated organisms contribute to the high risk of overwhelming sepsis in individuals with SCD. Penicillin prophylaxis and vaccination have significantly reduced the overall risk of Streptococcus pneumonia bacteremia, but an increased incidence of invasive pneumococcal infections with nonvaccine serotypes has been reported.28–30 Other encapsulated organisms are Haemophilus influenzae, Neisseria meningitidis, and Salmonella, and the latter has been known to cause osteomyelitis and pneumonia in SCD. Mycoplasma pneumoniae and Chlamydia pneumoniae should be considered in older children with infiltrates on chest radiograph. Viral infections (e.g., influenza and parvovirus B19) can result in severe morbidity.1,2,18,28,29,31,32
All SCD patients with fever greater than 38.3°C (101°F) must be evaluated to determine the risk of infection or sepsis; workup should include physical examination, complete blood count with reticulocyte count, blood culture, chest radiograph, urinalysis, and urine culture. Lumbar puncture may be needed, especially in young and toxic-appearing children. A low threshold for empiric therapy compared to that in the general population is recommended.1,28,31,33
Children with SCD may experience a severe complication due to infection that results in impaired bone marrow production of RBCs. An aplastic crisis is characterized by a decrease in the reticulocyte count and the rapid development of severe anemia (Table 82–2). The bone marrow becomes hypoplastic and is most often associated with a viral infection, particularly parvovirus B19.1,14,31
TABLE 82-2 Acute Sickle Cell Complications

Neurologic
Neurologic abnormalities and cognitive deficits are well documented in patients with SCD. Vasoocclusive processes can lead to cerebrovascular occlusion that manifests as signs and symptoms of overt stroke, such as headache, paralysis, aphasia, visual disturbances, facial droop, and convulsions. The risk of stroke is highest for HbSS and lowest for HbSβ+-thal. The occurrence of cerebral infarct in HbSS is 11% by age 20 years and 24% by age 45 years with a recurrence rate as high as 70% in 3 years. The highest risk occurs during the first decades, in particular ages 2 to 5. The risk is lowest before age 2 secondary to the protective effect of HbF. Ischemic strokes occur in 54% of cerebrovascular accidents with the highest risk before age 10 years and after 30 years of age; whereas hemorrhagic strokes are more common when patients are in their 20s and are associated with poor outcome.1,2,34
In addition to neurologic examination, evaluation of acute events include computed tomography (CT) scan and magnetic resonance imaging (MRI), magnetic resonance angiography for asymptomatic infarction, and transcranial Doppler (TCD) ultrasound to detect abnormal velocity and identify individuals at increased risk of stroke. In addition, electroencephalography (EEG) can be used if there is a history of seizure.1,2
About 10% to 30% of SCD who have HbSS with no prior history of stroke have been found to have changes on MRI of the brain consistent with infarction or ischemia. These “silent infarcts” can be associated with increased risk of stroke, decreased neurocognitive functions, behavioral changes, and poor academic performances. Finally, lower intelligence, visual-motor impairments, and neuropsychological dysfunctions have been reported in patients not affected by acute or silent strokes and are associated with severity of anemia.1,34–37
Acute Chest Syndrome
ACS is the second most common cause of hospitalization and responsible for about 25% of death among individuals with SCD. ACS is defined as a new pulmonary infiltrate associated with one or more of the following: cough, dyspnea, tachypnea, chest pain, fever, wheezing, and new-onset hypoxia. As many as one-half of individuals with SCD experience at least one episode of ACS. Risk factors for ACS and recurrence include young age (peak incidence between age 2 to 4 years), higher Hb, lower HbF, higher leukocytes, history of asthma or bronchial hyper-responsiveness, and smoke exposure. Genotype and haplotype also influence the occurrence. Patients with HbSS and HbSβ0-thal have higher incidence than those with HbSC and HbSβ+-thal. The prevalence is higher with African haplotypes than that of Saudi Arabia.1,25,38,39
The primary etiology for ACS is pulmonary vascular occlusion. The most common cause of ACS is infectious and may be of bacterial, viral, or mycoplasma in origin. Infections, fat emboli released from bone marrow during pain crisis, or direct adhesion of RBC to the pulmonary vasculature leads to the inflammation and injury of the lung. Predictors for respiratory failure include extensive lobar involvement, platelet count less than 200,000 cells/mm3 (200 × 109/L), and history of cardiac or neurologic complications. In addition to physical examinations, evaluations should include chest radiographs. In severe cases, CT scan, perfusion scintigraphy, transthoracic echocardiography, and bronchoscopy should also be considered. Pulmonary changes often involve the lower lobes of the lungs and may cause pleural effusions. Bilateral infiltrates or multiple lobe involvements may be an indication of poor prognosis. The mortality rates for ACS are less than 1%, 3%, and 9% for children up to 9 years old, children 10 to 19 years old, and adults, respectively. Pulmonary manifestations must be recognized early and managed aggressively as ACS can rapidly progress to pulmonary failure and death.25,39,40
Priapism
Stasis and sickling of erythrocytes within the sinusoids of the corpora cavernosa is the primary mechanism of priapism, a sustained painful erection. In recent years, a better understanding of pathophysiology of priapism has identified other mechanisms at the molecular level, such as NO and adenosine pathways. Stuttering priapism is repeated intermittent attacks up to several hours before remission; whereas ischemic priapism is a persistent painful erection greater than 4 hours and should be considered as an emergency. Thirty to 45% of males with SCD will present with at least one episode of priapism during their lifetime and the first episodes often occur during childhood. Impotence has been reported after repeated episodes and is directly related to the duration prior to treatment. ASPEN (association of sickle cell disease, priapism, exchange transfusion, and neurologic events) syndrome has occurred one to 11 days after partial exchange transfusion in males who presented with priapism. This syndrome can range from headaches and seizures to obtundation requiring ventilation.41–43
Sickle Cell Pain
Chronic hemolytic anemia in the SCD patient is periodically interrupted by acute episodes of pain and vasoocclusion, particularly in childhood (Table 82–2). Although fever, infections, dehydration, hypoxia, acidosis, and sudden temperature alterations can precipitate pain, multiple factors often contribute to its development.44
Sickle cell pain may be caused by bone or muscle infarction due to vasoocclusion and may affect a single site such as an arm, leg, or back or may involve multiple areas of the body. Acute painful episode is the most common reason of hospitalization. Individuals with HbSS disease experience frequent episodes of pain than those HbSC disease or some other variants.1,2,14 Biomarker for severity of pain during vasoocclusion episodes include C-reactive protein and lactate dehydrogenase (LDH).4,45 In addition, higher pain score at presentation and older age are predictors for admission and longer duration of hospitalization.46 Dactylitis (hand-and-foot syndrome) is a sub-type of sickle cell pain, occurring in infancy and early childhood and is characterized by redness and swelling of the dorsal aspects of the hands, feet, fingers, and toes. The episodes are painful but usually do not result in permanent damage.1
Splenic Sequestration
Splenic sequestration is the sudden massive enlargement of the spleen resulting from the sequestration of sickled RBCs in the splenic parenchyma (Table 82–2). Hematocrit and hemoglobin concentrations dramatically fall, with reticulocytosis and no evidence of marrow failure or accelerated hemolysis. Trapping of the sickled RBCs by the spleen also leads to a decrease in circulating blood volume, which can result in hypotension and shock. The condition is most often seen in infants and children because their spleens are intact, and can cause sudden death in young children. Splenic enlargement may also be acutely painful due to rapid capsular expansion. Over time, repeated splenic infarctions lead to autosplenectomy and the spleen can no longer become engorged; the incidence therefore declines as adolescence approaches.1,47
Chronic Complications
Pulmonary
Pulmonary hypertension as defined as an elevated tricuspid regurgitation (TR Jet) velocity has been reported to be a risk factor for death in adult patients with SCD.48 Diagnosis based on screening echocardiogram has been reported in children and adolescents, although its significance has not been established. Since recent studies have reported that screening echocardiogram alone has a high-false positive rate, pulmonary hypertension must be confirmed with right heart catheterization.48,49
The prevalence of asthma in children with SCD is similar to that of general population. However, asthma in individuals with SCD is associated with ACS and vasoocclusive pain episodes and increased mortality. Early screening for asthma starting at age 1 and pulmonary function test at least every 5 years starting at age 6 have been recommended. Medications such as inhaled corticosteroids and β-agonists for asthma exacerbation should be utilized for asthma control.1,27,48
Skeletal and Skin Diseases
Bone diseases are common in SCD and the vitamin D level has been suggested to be the biomarker.4 Osteonecrosis, particularly of the femoral or humeral heads, causes permanent damage and disability.14,50,51 Osteopenia associated with low bone formation has been reported in both males and females with SCD.52 Children with SCD also have an increased incidence of osteomyelitis; the organism most often responsible is Salmonella.31 In addition to necrosis of joints, chronic leg ulcers most commonly seen in the medial and lateral malleolus (ankles) can become a difficult and painful problem for adult. Ulcers are often seen after trauma or infection and are usually slow to heal.53
Ocular Manifestations
Ocular problems seen in patients with SCD include transient monocular blindness, visual field defects from retinal hemorrhage, retinal detachment, vitreous hemorrhage, venous microaneurysms, and neovascularization. The incidence of proliferative retinopathy in SCD patients varies from 5% to 10%. Vasoocclusion in the eye can occur as early as 20 months of age, and clinically detectable retinal diseases usually occur during adolescence and early adulthood. Despite the less systemic manifestations, individuals with HbSC develop serious retinal complications more often and earlier than those with HbSS. Annual examination with retinal evaluation is recommended for patients with SCD to prevent blindness from retinopathy and other complications.54
Gastrointestinal Diseases
Cholelithiasis is a common complication of SCD resulting from chronic hemolysis and increased bilirubin production, and leading to biliary sludge and/or stone formation. The risk of gallstones increases with age: 14% by age 10 years and 50% by age 22 years. Cholecystitis, exemplified by pain in the right upper quadrant, can be confused with an acute sickle pain episode in the abdomen. Cirrhosis occurred in 18% of young adults with SCD. Causes for development of chronic hepatic disease include repeated occlusion in the liver, iron overload, and hepatitis.1,14,55
Cardiac Diseases
Cardiovascular complications associated with anemia, including cardiac enlargement and various murmurs, can occur in patients with SCD. Patients experience various degrees of exertional dyspnea, tachycardia, and palpitation because of the decreased oxygen-carrying capacity of the blood. Left ventricular diastolic dysfunction has been reported in 18% of adults with SCD and is associated with increased mortality, especially in patients with pulmonary hypertension. Left ventricular stiffness and left ventricular hypertrophy have been reported, and the progression is speculated to lead to diastolic dysfunction later in life. Acute myocardial infarction in adults with SCD may be underrecognized due to the high incidence of sickle cell acute chest pain.56,57
Renal
Sickling in the renal medulla, an area with relative hypoxia and low pH, can lead to sickle cell nephropathy. Renal dysfunction in SCD begins during infancy evidenced by glomerular hyperfiltration. Other manifestations include inability to concentrate urine, hematuria, tubular acidosis, papillary necrosis, glomerulonephritis, microalbuminuria, and proteinuria. Enuresis, as a result of increased urine production, is a common complaint. Chronic renal failure has been reported in 30% of adults with SCD, and end-stage renal disease is associated with high mortality.2,14,58,59
Growth and Development
Delayed growth and sexual maturation are common in patients with SCD. Despite normal birth weight and length, growth retardation occurs between 6 months and 4 years with height, weight, and bone mass index being affected. The poor growth cannot be explained by nutritional factors alone. Alternations in growth factors as well as increased metabolic rate are factors contributing to the growth failure. Delay in puberty by 1 to 2 years is common in adolescents with SCD and fertility problems tend to occur more often in female SCD patients than in normal women.60,61
Psychiatric
Depression and anxiety are more common in children and adults with SCD than in the general population and have a significant impact on quality of life.62,63 Depression is associated with pain episodes in children with SCD. In addition, the level of depression has been reported to be associated with anxiety between mothers and children.64 DSM-IV psychiatric diagnosis in adolescents with SCD include attention-deficit-hyperactivity, oppositional defiant, conduct, major depressive, and anxiety disorders.65
Pregnancy
Pregnancy introduces an increased risk for the mother with SCD and for the fetus. Some women experience increased pain episodes during pregnancy and the anemia of SCD can lead to intrauterine growth retardation. Preterm labor and premature delivery are common in mothers with SCD, and the risk of spontaneous abortion is increased. The incidence of cesarean delivery and pregnancy-related complications are higher when compared to mothers who do not have SCD.14,66,67
TREATMENT
Sickle Cell Disease
Desired Outcomes
The goal of treatment is to reduce hospitalizations, complications, and mortality. Treatment for SCD involves the use of general measures to meet the unique demands for increased erythropoiesis. Additional interventions can be aimed at preventing or treating complications of the disease. When an acute complication occurs, the type and severity of the episode determine the appropriate therapeutic plan.
With availability of public health programs and comprehensive care, most children survive through childhood.12,26,68 The median survival rate is estimated to be 42 years for males and 48 years for females for HbSS, and 60 years for males and 68 years for females for HbSC.12 With increased life expectancy, management of SCD should include assessment of health-related quality of care in both adults and children.
Patients with SCD require lifelong multidisciplinary care. All patients with SCD should receive regularly scheduled comprehensive medical evaluations. Because of the complexity of the disease, a multidisciplinary team is needed to provide high-quality medical care, education, counseling, and psychosocial support. Appropriate comprehensive care can have a positive impact on both longevity and quality of life. This care includes the use of evidence-based treatment combining general symptomatic supportive care, preventative medical therapies, and more specific disease-modifying therapies aimed at altering hematologic capacity and function.
Routine Health Maintenance
Immunizations
Administration of routine immunizations is crucial preventive care in managing SCD. Impaired splenic function increases susceptibility to infection by encapsulated organisms, particularly Pneumococci. Prior to the routine use of penicillin prophylaxis and the development of pneumococcal vaccines, invasive pneumococcal disease was 20- to 100-fold more common in children with SCD than in healthy children. Reduced mortality has been associated with the introduction of pneumococcal vaccines.26,68
Two different pneumococcal vaccines are available. The 13-valent pneumococcal conjugate vaccine (PCV13; Prevnar®) induces good antibody responses in infants and children less than 2 years of age. Immunization with the PCV13 is recommended for all children, regardless of SCD status, younger than 24 months of age. Infants should receive the first dose after 6 weeks of age. Two additional doses should be given at 2-month intervals, followed by a fourth dose at age 12 to 15 months. The 23-valent pneumococcal polysaccharide vaccine (PPSV 23; Pneumovax®23) is recommended for all children with functional or acquired asplenia but must be given after 2 years of age because of poor antibody response. To cover different serotypes, PPSV 23 should be given starting at 2 years of age, and be administered 2 months after the last dose of the 7-valent pneumococcal conjugate vaccine (PCV7)/PCV13. A booster dose of PPSV 23 is recommended 5 years after the first dose. The recommended immunization schedule and catch-up schedule for PCV13 and PPV 23 are presented in Table 82–3. For children ages 2 to 18 years with SCD who did not receive PCV-13 during their routine immunization, a single dose of PCV-13 is recommended.69
TABLE 82-3 Pneumococcal Immunizations for Children with Sickle Cell Disease