Whereas the sex chromosomes play a determining role in specifying chromosomal and gonadal sex, a number of genes located on both the sex chromosomes and the autosomes are involved in sex determination and subsequent sexual differentiation. In most instances, the role of these genes has come to light as a result of patients with various conditions known as disorders of sex development, and many of these are discussed later in this chapter.
The Y Chromosome
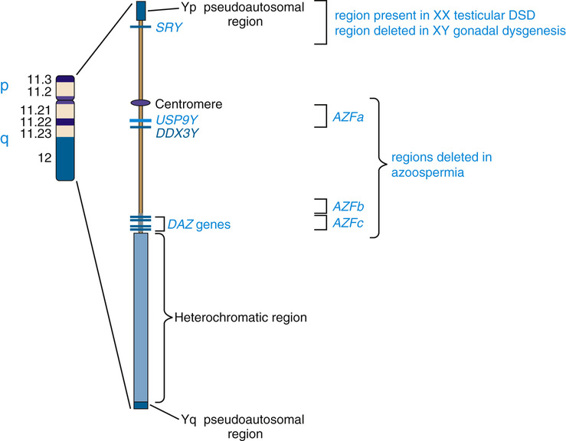
The structure of the Y chromosome and its role in sex development has been determined at both the molecular and genomic levels (Fig. 6-9). In male meiosis, the X and Y chromosomes normally pair by segments at the ends of their short arms (see Chapter 2) and undergo recombination in that region. The pairing segment includes the pseudoautosomal region of the X and Y chromosomes, so called because the X- and Y-linked copies of this region are essentially identical to one another and undergo homologous recombination in meiosis I, like pairs of autosomes. (A second, smaller pseudoautosomal segment is located at the distal ends of Xq and Yq.) By comparison with autosomes and the X chromosome, the Y chromosome is relatively gene poor (see Fig. 2-7) and contains fewer than 100 genes (some of which belong to multigene families), specifying only approximately two dozen distinct proteins. Notably, the functions of a high proportion of these genes are restricted to gonadal and genital development.
Embryology of the Reproductive System
The effect of the Y chromosome on the embryological development of the male and female reproductive systems is summarized in Figure 6-10. By the sixth week of development in both sexes, the primordial germ cells have migrated from their earlier extraembryonic location to the paired genital ridges, where they are surrounded by the sex cords to form a pair of primitive gonads. Up to this time, the developing gonad is ambipotent, regardless of whether it is chromosomally XX or XY.
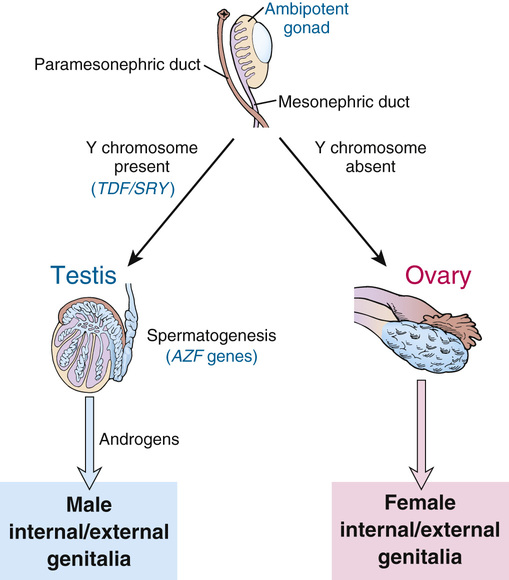
Development into an ovary or a testis is determined by the coordinated action of a sequence of genes in finely balanced pathways that lead to ovarian development when no Y chromosome is present but tip to the side of testicular development when a Y is present. Under normal circumstances, the ovarian pathway is followed unless a particular Y-linked gene, originally designated testis-determining factor (TDF), diverts development into the male pathway.
If no Y chromosome is present, the gonad begins to differentiate to form an ovary, beginning as early as the eighth week of gestation and continuing for several weeks; the cortex develops, the medulla regresses, and oogonia begin to develop within follicles (see Fig. 6-10). Beginning at approximately the third month, the oogonia enter meiosis I, but (as described in Chapter 2) this process is arrested at dictyotene until ovulation occurs many years later.
In the presence of a normal Y chromosome (with the TDF gene), however, the medullary tissue forms typical testes with seminiferous tubules and Leydig cells that, under the stimulation of chorionic gonadotropin from the placenta, become capable of androgen secretion (see Fig. 6-10). Spermatogonia, derived from the primordial germ cells by successive mitoses, line the walls of the seminiferous tubules, where they reside together with supporting Sertoli cells, awaiting the onset of puberty to begin spermatogenesis.
While the primordial germ cells are migrating to the genital ridges, thickenings in the ridges indicate the developing genital ducts, the mesonephric (also called wolffian) and paramesonephric (also called müllerian) ducts, under the influence of hormones produced by specific cell types in the developing gonad. Duct formation is usually completed by the third month of gestation.
In the early embryo, the external genitalia consist of a genital tubercle, paired labioscrotal swellings, and paired urethral folds. From this undifferentiated state, male external genitalia develop under the influence of androgens, beginning at around 12 weeks of gestation. In the absence of a testis (or, more specifically, in the absence of androgens), female external genitalia are formed regardless of whether an ovary is present.
SRY is the Major Testis-Determining Gene
The earliest cytogenetic studies established the male-determining function of the Y chromosome. In the ensuing three decades, chromosomal and genomic analysis of individuals with different submicroscopic abnormalities of the Y chromosome and well-studied disorders of sex development allowed identification of the primary testis-determining region on Yp.
Whereas the X and Y chromosomes normally exchange in meiosis I within the Xp/Yp pseudoautosomal region, in rare instances, genetic recombination occurs outside of the pseudoautosomal region (Fig. 6-11). This leads to two rare but highly informative abnormalities—males with a 46,XX karyotype and females with a 46,XY karyotype—that involve an inconsistency between chromosomal sex and gonadal sex, as we will explore in greater detail later in this chapter.
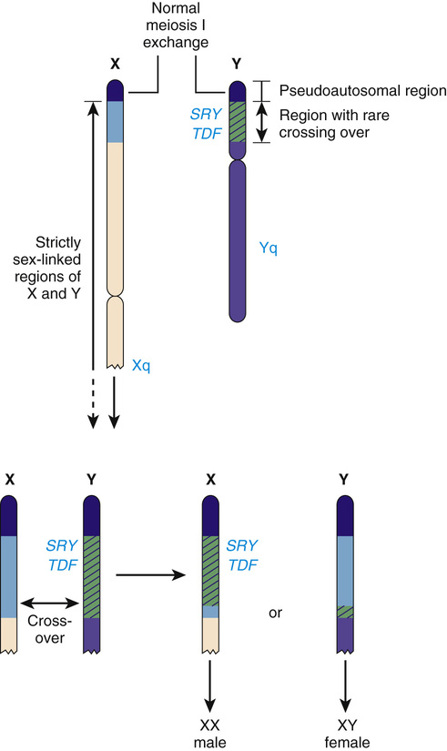
The SRY gene (sex-determining region on the Y) lies near the pseudoautosomal boundary on the Y chromosome. It is present in many males with an otherwise normal 46,XX karyotype (Case 41) and is deleted or mutated in a proportion of females with an otherwise normal 46,XY karyotype, thus strongly implicating SRY in normal male sex determination (see Fig. 6-11). SRY is expressed only briefly early in development in cells of the germinal ridge just before differentiation of the testis. SRY encodes a DNA-binding protein that is likely to be a transcription factor, which up-regulates a key autosomal gene, SOX9, in the ambipotent gonad, leading ultimately to testes differentiation. Thus, by all available genetic and developmental criteria, SRY is equivalent to the TDF gene on the Y chromosome. If SRY is absent or not functioning properly, then the female sex differentiation pathway ensues (see Fig. 6-10).
Although there is clear evidence demonstrating the critical role of SRY in normal male sexual development, the presence or absence of SRY/TDF does not explain all cases of abnormal sex determination. Other genes are involved in the sex determination pathway and are discussed later in this chapter.
Y-Linked Genes in Spermatogenesis
The prevalence of Y chromosome deletions and microdeletions in the general male population is reported to be approximately 1 in 2000 to 3000 males. However, microdeletions in the male-specific portion of Yq are found in a significant proportion of men with low sperm count, ranging from cases of nonobstructive azoospermia (no sperm detectable in semen) to severe oligospermia (<5 million/mL; normal range, 20 to 40 million/mL). These findings suggest that one or more genes, termed azoospermia factors (AZF), are located on the Y chromosome, and three such regions on Yq (AZFa, AZFb, and AZFc) have been defined (see Fig. 6-9).
Genomic analysis of these microdeletions led to identification of a series of genes that appear to be important in spermatogenesis. For example, the 3.5-Mb-long AZFc deletion region contains seven different families of genes that are expressed only in the testis, including four copies of the DAZ genes (deleted in azoospermia) that encode nearly identical RNA-binding proteins expressed only in the premeiotic germ cells of the testis. De novo deletions of AZFc arise in approximately 1 in 4000 males and account for approximately 12% of azoospermic males and approximately 6% of males with severe oligospermia. Deletion of only two of the four DAZ genes has been associated with milder oligospermia. Similar to the other genomic disorders described earlier in this chapter, they are mediated by recombination between segmentally duplicated sequences (see Table 6-3). AZFa and AZFb deletions, although less common, also involve recombination. The Yq microdeletions are not syndromic, however; they are responsible only for a defect in spermatogenesis in otherwise normal males. The explanation is that all of the genes involved in the AZF deletions are expressed only in the testis and have no functions in other tissues or cell types.
Overall, approximately 2% of otherwise healthy males are infertile because of severe defects in sperm production, and it appears likely that de novo deletions or mutations of genes on Yq account for a significant proportion of these. Thus men with idiopathic infertility should be karyotyped, and Y chromosome molecular testing and genetic counseling may be appropriate before the initiation of assisted reproduction by intracytoplasmic sperm injection for such couples, mostly because of the risk for passing a Yq microdeletion responsible for infertility to the infertile couple’s sons.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


