Secondary and Tertiary Hyperparathyroidism
Kaare J. Weber
Shalini Arora
Introduction
Secondary hyperparathyroidism is a physiologic response to a defect in calcium homeostasis. Causes are broad and can include genetic, gastrointestinal, vitamin D-related, and renal-related causes (Table 1). Nearly all patients with chronic renal failure develop some degree of secondary hyperparathyroidism. Therefore, secondary hyperparathyroidism in the setting of chronic renal failure remains a challenge to both nephrologists and endocrine surgeons. Previous estimates suggested that 5% of the renal failure population undergo surgery for hyperparathyroidism every year; however, with advances in medical management, that number is now thought to be 1% per year, with an overall estimate of 10% eventually undergoing parathyroidectomy.
Etiology
Calcium regulation responds to both parathyroid hormone released by the parathyroid glands and active vitamin D (1,25-dihydroxyvitamin D3) produced by the kidney. The main target organs of these two hormones are the bones, kidneys, and intestines. The exact etiology of secondary hyperparathyroidism remains unclear but
is thought to be multifactorial. Decreased active vitamin D production, hypocalcemia, and phosphate retention all contribute to the development of secondary hyperparathyroidism. As glomerular filtration rate declines, circulating levels of active vitamin D begin to decrease due to diminished hydroxylation of 25-hydroxyvitamin D to the active 1,25-dihydroxyvitamin D. As a result, a decrease in absorption of calcium from the gut leads to hypocalcemia. Hypocalcemia combined with hyperphosphatemia due to impaired excretion by the kidneys, leads to stimulation of the parathyroid glands to release parathyroid hormone. The result is hyperplasia of all parathyroid glands due to polyclonal cell proliferation and compensatory increase of parathyroid hormone in attempts to raise serum calcium levels. Continued stimulation of the parathyroid glands leads to formation of monoclonal cell proliferation within the hyperplastic tissue and can result in the development of nodular hyperplasia. The parathyroid cells of nodular hyperplasia have been shown to have decreased expression of both the vitamin D and calcium-sensing receptors.
is thought to be multifactorial. Decreased active vitamin D production, hypocalcemia, and phosphate retention all contribute to the development of secondary hyperparathyroidism. As glomerular filtration rate declines, circulating levels of active vitamin D begin to decrease due to diminished hydroxylation of 25-hydroxyvitamin D to the active 1,25-dihydroxyvitamin D. As a result, a decrease in absorption of calcium from the gut leads to hypocalcemia. Hypocalcemia combined with hyperphosphatemia due to impaired excretion by the kidneys, leads to stimulation of the parathyroid glands to release parathyroid hormone. The result is hyperplasia of all parathyroid glands due to polyclonal cell proliferation and compensatory increase of parathyroid hormone in attempts to raise serum calcium levels. Continued stimulation of the parathyroid glands leads to formation of monoclonal cell proliferation within the hyperplastic tissue and can result in the development of nodular hyperplasia. The parathyroid cells of nodular hyperplasia have been shown to have decreased expression of both the vitamin D and calcium-sensing receptors.
Table 1 Differential Diagnosis of Secondary Hyperparathyroidism | ||||||||
|---|---|---|---|---|---|---|---|---|
|
Prolonged stimulation of the parathyroid glands may result in autonomous production of parathyroid hormone even after removal of the physiologic stimulus, leading to tertiary hyperparathyroidism. This condition occurs in up to 30% of patients with chronic renal failure after kidney transplantation. It can, however, occur in any patient with secondary hyperparathyroidism with long-standing hypocalcemia, and should be suspected in patients with renal hyperparathyroidism who become hypercalcemic. Etiology is thought to be due to the loss of calcium-sensing and vitamin D receptors on the parathyroid glands resulting in elevated parathyroid hormone and normal or elevated serum calcium levels. Tertiary hyperparathyroidism resembles primary hyperparathyroidism because of the autonomously functioning parathyroid glands from a monoclonal expansion of adenomatous like tissue. Although it most commonly occurs due to hyperplasia of all four glands, there are reports of single and double glands causing the hyperparathyroid state in 2.6 to 32% of patients.
Secondary hyperparathyroidism can lead to a spectrum of bone disease referred to renal osteodystrophy. There are four main types of bone diseases seen in renal failure patients and include osteitis fibrosa cystica, adynamic bone disease, osteomalacia, and mixed uremic osteodystrophy in which elements of both high and low bone turnover are seen. Subclinical changes in mineral metabolism and bone structure begin early in the course of kidney disease but signs and symptoms of bone disease typically do not occur until the patient is already on dialysis. Patients can present with weakness, bone pain, myopathy, and fractures.
In addition to bone disease, uremic pruritus can be quite debilitating, impacting greatly on quality of life. Etiology is multifactorial but is thought be due to changes in calcium and phosphate metabolism with elevated calcium/phosphate product, increased parathyroid hormone, anemia, and high aluminum levels.
Persistent hyperparathyroidism can also lead to calcium phosphate precipitation resulting extraosseous calcification of the joints, soft tissues, and viscera. High parathyroid hormone levels can induce vascular calcifications, including coronary artery calcification.
Renal hyperparathyroidism is associated with an increase in cardiovascular morbidity and mortality. Mechanisms are not clear but are thought to be due to derangements in calcium and phosphate metabolism leading to accelerated vascular calcification increasing adverse cardiovascular events. Hyperphosphatemia has also been identified as an independent risk factor for decline in renal function and higher mortality.
A rare but life-threatening complication of secondary hyperparathyroidism is calciphylaxis. Calciphylaxis is an entity of excessive calcium deposition in the microvasculature, leading to ischemia and ulcers in atypical areas of the body (Fig. 1). The ulcers typically do not respond to debridement and local wound care. Often these wounds become super-infected, resulting in sepsis and death.
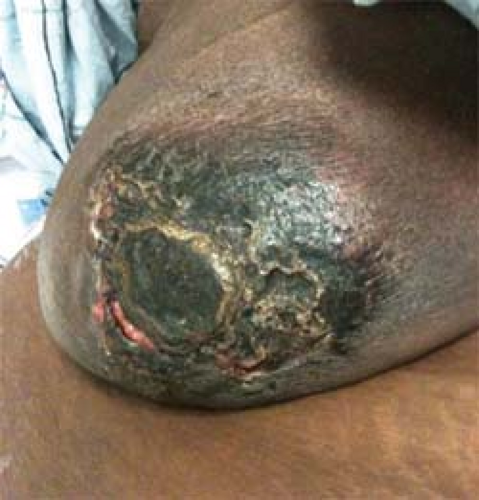 Fig. 1. Calciphylaxis right breast/nipple–areolar complex. |
Although most patients with tertiary hyperparathyroidism remain asymptomatic, those presenting with symptoms mirror that of primary hyperparathyroidism. These include nephrolithiasis, peptic ulcer disease, pancreatitis, and bone disease. Following transplant, high parathyroid hormone levels stimulate the transplanted kidney to produce activated Vitamin D. This in turn results in increased bone turnover and absorption of calcium from the gut leading to hypercalcemia. In patients who remain asymptomatic, calcium levels over 12 mg/dL may cause graft deterioration.
Medical Management
Medical therapy of secondary hyperparathyroidism is based upon knowing the major factors contributing to excess parathyroid hormone release, including vitamin D deficiency, hypocalcemia, and hyperphosphatemia. The current management of secondary hyperparathyroidism involves some combination of calcium and vitamin D supplementation, phosphate binders, and a calcimimetic. Calcimimetics have been added to the nephrologists’ armamentarium more recently. They are called “calcimimetic” because they mimic the effects of extracellular calcium on parathyroid glands. They act by occupying the calcium-sensing receptor on the parathyroid glands, thus down-regulating parathyroid hormone release. Cinacalcet has been shown to improve parathyroid hormone, calcium, and phosphate levels with early evidence showing reduced risk of parathyroidectomy, fracture, and cardiovascular events in renal failure patients. Prior to the introduction of calcimimetics, <5% of the renal population came to surgery for symptomatic secondary hyperparathyroidism.
Current estimates suggest that only 1% of the renal failure population will require parathyroidectomy with the introduction of calcimimetics.
Current estimates suggest that only 1% of the renal failure population will require parathyroidectomy with the introduction of calcimimetics.
Tertiary hyperparathyroidism is characterized by hypercalcemia and hypophosphatemia and therefore does not respond to the above regimen for secondary hyperparathyroidism. Minimal evidence exists that calcimimetics may benefit patients suffering from tertiary hyperparathyroidism and therefore calcimimetics are currently not approved for this entity. Surgery remains the only therapeutic option for these patients.
Indications for surgery for secondary hyperparathyroidism can be broken down into both biochemical and clinical indications. Guidelines have been developed to improve clinical outcomes for patients suffering from renal hyperparathyroidism. The Kidney Disease Outcomes Quality Initiative (KDOQI) specifies targets for calcium and bone mineral metabolism to slow disease progression and minimize complications (Table 2). Surgery is often employed when medical management “failed” to meet these guidelines. However, randomized controlled trials do not exist indicating when parathyroidectomy should be performed. Parathyroidectomy is generally recommended when serum levels of intact parathyroid hormone are >800 pg/mL in association with hypercalcemia and/or hyperphosphatemia. Also a calcium/phosphate product of >55 implies failed medical therapy.
Size of parathyroid glands is a relative indication for surgery. Glands with an estimated volume of >500 mm3 or diameter greater than 1 cm on ultrasound are likely to have developed nodular hyperplasia. This is based on the belief that nodular hyperplasia of secondary hyperparathyroidism has diminished expression of both vitamin D and calcium-sensing receptors leading to a diminished response to medical therapy.
Regardless of absolute laboratory values or size of the parathyroid glands, surgery is often indicated with the onset of symptoms. These include bone and joint pain, muscle weakness, and itching. Moreover, bone loss, fractures, and extra-skeletal calcifications are indications for surgery.
Table 2 KDOQI Bone Mineral Metabolism Targets | |||
|---|---|---|---|
|
Table 3 Indications for Surgery in Tertiary Hyperparathyroidism | ||||||
|---|---|---|---|---|---|---|
|
There is an ongoing controversy on the approach to calciphylaxis. However, because mortality rates can be as high as 90%, calciphylaxis is best managed by controlling secondary hyperparathyroidism with parathyroidectomy. Once the biochemical abnormalities are corrected, the ischemia and ulcers of calciphylaxis are then allowed to heal with ongoing local wound care.
Similar to secondary hyperparathyroidism, tertiary hyperparathyroidism lacks clear indications for surgery based on randomized controlled studies. Elevated parathyroid hormone levels are seen in over 25% of patients 1 year after kidney transplantation; however, up to 5% of posttransplant patients will ultimately need parathyroidectomy. Proposed indications for surgery are also based on biochemical and clinical criteria (Table 3). While the standard delay of surgery is 1 year, surgery can be performed at 3 months because most patients should recover normal parathyroid function by this time.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


