(1)
BIOTEC, Biophysics, Technische Universität Dresden, Dresden, Germany
(2)
Cellular and Molecular Biophysics, Max Planck Institute of Biochemistry, Martinsried, Germany
Abstract
Scanning fluorescence correlation spectroscopy (SFCS) with a scan path perpendicular to the membrane plane was introduced to measure diffusion and interactions of fluorescent components in free-standing biomembranes. Using a confocal laser scanning microscope (CLSM), the open detection volume is repeatedly scanned through the membrane at a kHz frequency. The fluorescence photons emitted from the detection volume are continuously recorded and stored in a file. While the accessory hardware requirements for a conventional CLSM are minimal, data evaluation can pose a bottleneck. The photon events must be assigned to each scan, in which the maximum signal intensities have to be detected, binned, and aligned between the scans, in order to derive the membrane-related intensity fluctuations of one spot. Finally, this time-dependent signal must be correlated and evaluated by well-known FCS model functions. Here we provide two platform-independent, open source software tools (PyScanFCS and PyCorrFit) that allow to perform all of these steps and to establish perpendicular SFCS in its one- or two-focus as well as its single- or dual-color modality.
Key words
Scanning fluorescence correlation spectroscopy (SFCS)Fluorescence correlation spectroscopy (FCS)Fluorescence cross-correlation spectroscopy (FCCS)Membrane diffusionGiant unilamellar vesicles (GUV)DiffusionProtein–protein interactionLigand binding1 Introduction
In recent years, fluorescence correlation spectroscopy (FCS) has become a well-established method in biomedical research [1, 2]. In addition to confocal laser scanning microscopy (CLSM), where the spatial fluorescence intensity distribution is sampled, FCS records the time-dependent intensity fluctuations originating from a microscopic spot. With sufficient sensitivity and time resolution of the detectors, the signal fluctuations will reflect not only instrumental noise but also the stochastic Brownian motion of the fluorescent molecules performing random walks through the illuminated, open detection volume. Thus, the time-dependent signal contains information about molecular properties which can be extracted and analyzed (see Chapter 24).
The average time required by molecules to “walk” through the detection volume (diffusion time) depends on its shape and size and is therefore characteristic for the optical setup. The mobility of the molecules depends on their shape and size, as well as the viscosity of the surrounding medium (see Chapter 24). Because the viscosity of a lipid bilayer or a cellular plasma membrane is about 100 times larger than that of the aqueous media, the diffusion times are slowed down to the same degree. In addition, the diffusion is confined to a very thin membrane plane of 5–10 nm thickness. Using diffraction limited illumination, lateral, two-dimensional diffusion times of lipids are typically in the millisecond (ms) regime and single-pass transmembrane receptors are even 20–50 times slower.
Perpendicular scanning (SFCS) was introduced to measure mobility and interactions in the lipid bilayer of giant unilamellar vesicles (GUVs) [3, 4]. Slower diffusion of fluorescent molecules in combination with small concentrations produces rare events with longer lifetime, and therefore, it is sufficient to collect data with an appropriate sampling frequency. In perpendicular line SFCS [3, 5, 6], this is achieved by guiding the focused laser beam repeatedly along a straight line with kHz frequency perpendicular through the membrane plane (Fig. 1). Scanning is performed by means of the conventional control software of a CLSM. At all times, the emitted photons are collected with single-photon counting detectors, e.g., avalanche photo diodes (APDs). The signal is digitalized by a separate device and stored in a file. Here we used a USB connected hardware correlator; however, other time-correlated single-photon counting (TCSPC) cards may also be suitable. Since the file contains all photon incidents along the scan path, the signal originating from the spot where the scan path crosses the membrane plane must be extracted post-measurement.
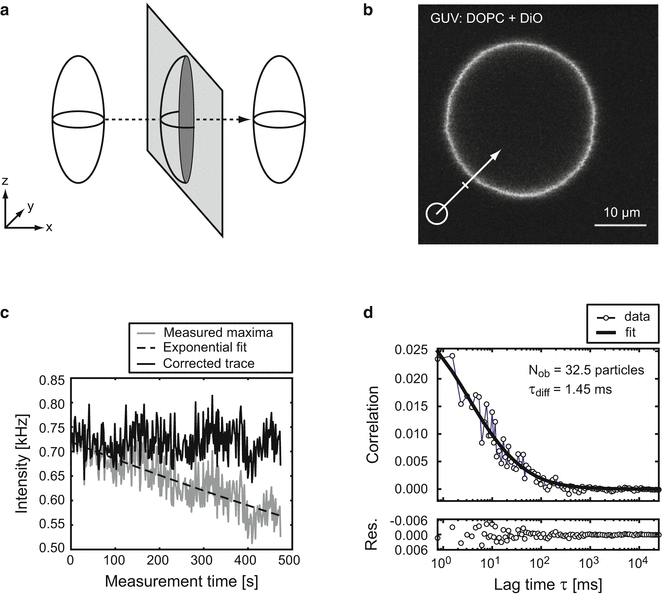
Fig. 1
Single-focus perpendicular SFCS. (a) Graphical representation of the open detection volume as an ellipsoid, stretched along the optical axis z. Lateral movement on a perpendicular scan path (dashed arrow) through a two-dimensional membrane plane (light gray) creates an elliptical, two-dimensional detection area (dark gray). (b) Equatorial confocal image of a GUV composed of DOPC containing 0.007 % of the fluorescent lipid DiO. The scan path (white arrow) was positioned perpendicular to the membrane plane at a 45° angle. (c) Intensity trace of the maxima (gray) along each scan is plotted against the measurement time (500 s = 6.5 × 105 scans with a cycle time 0.768 ms). During the measurement, the intensity dropped exponentially (dashed) by about 25 % due to bleaching, and was back-corrected (black) [6]. The magnitude of the intensity values (here about 0.75 kHz) depends on the exact procedure to assign membrane-related fluorescence, in particular the number of bins used to sample each line scan (here 70) and the number of bins assigned to the membrane area (here 7: 1 maximum plus 6 adjacent bins). (d) Autocorrelation curve of the corrected intensity trace from (c) was fitted with a model function for two-dimensional diffusion through an elliptical Gaussian shaped detection volume (Eq. 2)
For this purpose, we developed a program (PyScanFCS). The correct periodicity (scan cycle time) corresponding to the repetition rate of each scan is determined by means of Fourier analysis of the photon arrival times. For visual inspection, the aligned scan intensities are displayed in a kymograph containing the number of scans on the x-axis and the intensity along the scan path on the y-axis. The time range displaying homogeneous intensity can be manually selected. The number of bins along the scan path containing membrane-related intensity can be defined and corrected for systematic bleaching during the course of the measurement (typically several minutes). Finally, the resulting intensity trace is time-correlated by a multiple τ algorithm. To evaluate the correlation data, the curves can be analyzed with a second program (PyCorrFit) written for fitting different model functions containing molecular parameters like diffusion times, diffusion coefficients, and particle numbers.
Compared to single-point confocal FCS, perpendicular SFCS has several advantages when applied to membranes [5, 7]. Free-standing membranes are usually large structures showing slow thermal motion like undulations. During the measurement, the vesicle or the cell may move or change shape. This is a problem for conventional single-point confocal FCS, because the detection volume is fixed in space; even minor displacements of the fluorescent membrane with respect to a fixed detection volume cause a detrimental bias in correlation curves. Because in SFCS the maximum intensity on each path is determined post-measurement, such positional instabilities can be corrected. Additionally, the laser focus will be outside the membrane most of the time and therefore the amount of bleaching is reduced. However, a potential disadvantage of the SFCS approach is a low average signal-to-noise ratio and accordingly prolonged measurement times.
We demonstrate how to obtain and evaluate perpendicular SFCS data by measuring the diffusion of the 488 nm excitable, fluorescent lipid 3,3′-dioctadecyloxacarbocyanine (DiO) in GUVs composed of 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC). Scanning is either performed on a single line (single-focus SFCS) or on a frame composed of two lines (two-focus SFCS). Two-focus SFCS takes advantage of the fixed distance between the scan paths to obtain calibrated diffusion coefficients [8]. Additional scan modalities like dual-color scanning fluorescence cross-correlation spectroscopy (SFCCS) are briefly outlined. Dual-color SFCCS is a powerful approach to address interactions between differently labelled chemical species in biomembranes [9, 10].
2 Materials
2.1 Preparation of GUVs
1.
10 ml 200 mM sucrose in H2O.
2.
10 ml 200 mM glucose in H2O.
3.
Preparation of lipid mixture: Take DOPC stock solution (20 mg/ml in chloroform) and DiO (5 μM in chloroform) from −20 °C and dilute with chloroform to final concentration of 2 mg/ml DOPC and a relative concentration of DiO in DOPC of 0.007 mol% (Avanti Polar Lipids, Alabaster, Alabama, USA). 5–10 μl is used to prepare GUVs in the Teflon chamber.
4.
Bovine serum albumin (BSA), 2 mg/ml in H2O (Sigma-Aldrich, Munich, Germany).
5.
Vacuum chamber for drying samples (Vacuubrand, Wertheim, Germany).
7.
Electrical sine wave generator (TTi TG315 function generator, Huntingdon, Cambridgeshire, UK).
2.2 Microscopy
1.
8-well chambered cover slides (LabTek II, #1.5, Nunc, Thermo Scientific, USA).
2.
Diffusion standard to calibrate the detection volume: hydrolyzed AlexaFluor 488 succinimidylester (Life technologies/Molecular Probes, USA). Prepare a 25 nM solution in 10 mM Tris-HCl, pH 8.
3.
Purified enhanced green fluorescent protein (EGFP) (CatNo.:#4999-100, BioVision, USA).
2.3 Hardware for SFCS Data Acquisition
CLSM setups equipped with photon counting detectors (e.g., a LSM510 with accessory Confocor3 unit, Zeiss, Germany) offer direct access to the signal by means of BNC-connected coaxial cables attached to the APDs. If the CLSM does not possess photon counting detectors, one can use the fiber-out exit and feed the signal into external APDs [3]. For this, one needs:
1.
APDs (Perkin-Elmer Optoelectronics, USA).
2.
Multimode fibers with MM connectors, ASF105/125Y (Thorlabs, Newton, New Jersey, USA).
3.
Positioning system (miniature xyz stage) with FC adapter to align the optical fiber (Owis, Staufen, Germany).
4.
USB connectable hardware correlator to record the photon history (Model Flex02-12D/B, www.correlator.com, USA/China) (see Note 2).
2.4 Software for SFCS Data Acquisition
The data processing software (PyScanFCS and PyCorrFit) has been tested on Windows XP/7 and Ubuntu Linux (see Note 3).
1.
Software for photon history recording, Photon.exe (shipped with the Flex correlator Flex02-12D/B, www.correlator.com).
2.
Program to generate correlation curves from the photon history file PyScanFCS (free download at http://pyscanfcs.craban.de). Due to constraints in memory of 32-bit systems, we recommend to use a 64-bit computer with either Windows 7 or an Ubuntu AMD64 operating system.
3.
Fitting program to extract relevant parameters from correlation curves, PyCorrFit (free download at http://pycorrfit.craban.de).
3 Methods
3.1 Preparation of GUVs by Electroformation
1.
Clean the cap and reservoir of the Teflon chamber two times with 80 % ethanol and with hot tap water.
2.
Rinse the Teflon container and the cap with distilled water and dry both with pressurized air.
3.
Using a pipette, distribute a total 5 μl of the 2 mg/ml lipid–dye mixture evenly on both platinum electrodes pointing towards the inside of the chamber.
4.
To remove the chloroform, let the lipid mixture dry in air for 2 min and further in a vacuum chamber for 10 min.
5.
Fill the Teflon container with 300 μl of 200 mM sucrose solution and close the lid. The platinum electrodes should be completely covered by sucrose solution.
6.
Apply an alternating electric field of 2.15 V at 10 Hz for 1.5 h. During this phase, the GUVs form at the electrodes.
7.
Decrease the frequency to 2 Hz for another 20 min to allow the GUVs to detach from the electrodes.
8.
Incubate the Labtek chamber with 2 mg/ml bovine serum albumin (BSA) for 20 min.
9.
Wash the Labtek chamber five times with 200 mM glucose solution.
10.
Fill the Labtek chamber with 400 μl of 200 mM glucose solution.
11.
Add 50 μl of the GUV-sucrose solution. Cut off the tip when pipetting GUVs to prevent them from bursting due to shear forces (see Note 4).
3.2 Define the Microscope Settings for SFCS
3.2.1 Calibration with Alexa488 Solution
1.
Prepare the 8-well Labtek containing a 1–50 nM Alexa488 solution and the GUV solution in adjacent wells. Clean the glass bottom with ultra pure ethanol and mount on the stage.
2.
Define settings for excitation and detection (e.g., a dichroic 488/633 followed by BG35 to eliminate scattered UV light and a cleaning band pass (505–560) for Alexa488 and DiO emission).
3.
Set the CLSM to measure in a single spot (with LSM510 and external APDs this is possible by selecting scan mode “Spot” and performing a time series).



