Chapter 13 Scaffolds
Small globular proteins as antibody substitutes
Scaffolds
Monoclonal antibodies (mAbs) are an increasingly important class of therapeutic agents as discussed in preceding chapters. Over 25 recombinant antibodies are currently on the market, the top 10 best-selling of these products earned over $32 billion in global sales during 2008, and five mAb products (rituximab, infliximab, bevacizumab, trastuzumab and adalimumab) were included in the top 15 best-selling prescription drugs for that year (Reichert, 2010). The future prospects for therapeutic antibodies are clearly bright, the annual growth rate estimated to be 12% during 2010–2012. However, even antibody blockbusters suffer from certain drawbacks: human immunoglobulin G (IgG) antibody is a complex molecule composed of four protein chains with attached carbohydrates. A full-size IgG antibody is a Y-shaped complex of two identical light chains, each with a variable and constant domain, and two heavy chains, each with one variable and three constant domains (Fig. 13.1H). Therefore, full-size IgGs are always bivalent molecules and elicit effector functions through their Fc part, which may not be ideal for certain applications. With regards to expression, there is the requirement for a rather expensive mammalian cell production system and, in addition, antibodies depend on disulfide bonds for stability. Through the versatile techniques of genetic engineering, smaller antibody fragments have been produced to overcome some of the limitations of full-size IgGs (Holliger and Hudson, 2005), but some antibody fragments tend to aggregate and display limited solubility. Finally, the success, and consequently the extensive use, of antibodies has led to a complicated patent situation for antibody technologies and applications. Therefore, several research groups and small and medium-sized companies have recently focused on the development of small globular proteins as scaffolds for the generation of a novel class of versatile binding proteins.
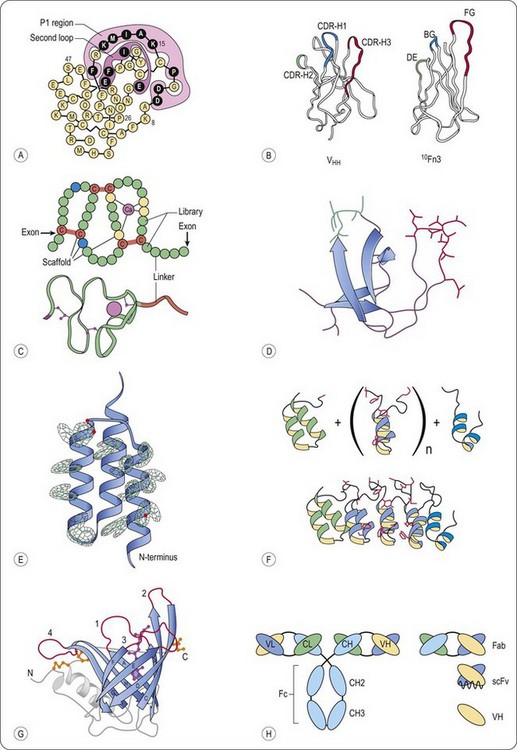
Fig. 13.1 Schematic representation of representative scaffolds. A, Kunitz type domain (LAC–D1) (Ley et al., 1996). Varied positions are depicted in black, the P1 and second loop positions are enclosed. B, Structural comparison of a llama VHH domain and the wild-type human 10Fn3 domain (Xu et al., 2002). Despite the lack of significant sequence identity, both domains fold into similar beta sheet sandwiches. The CDRs of the VHH domain and the residues randomized in the 10Fn3 domain are shown in color. C, Fixed and variable positions of the A-domain library, as well as the disulfide topology, are indicated (Silverman et al., 2005). Each circle represents an amino acid position. Calcium-coordinating scaffold residues are yellow, structural scaffold residues are blue, cysteine residues are red and variable positions are green. A ribbon diagram of a prototypical A-domain structure is included (Protein Data Bank entry 1AJJ). D, Fyn SH3 wt protein structure (Protein Data Bank entry 1M27) The RT-Src loop is in red, and the n-Src loop is in green. E, Electron density for all 13 mutated residues in the affibody (Hogbom et al., 2003). For clarity, only the electron density around the side chains is displayed. F, Schematic representation of the library generation of designed ankyrin repeat proteins (Binz et al., 2004). (upper part) Combinatorial libraries of ankyrin repeat proteins were designed by assembling an N-terminal capping ankyrin (green), varying numbers of the designed ankyrin repeat module (blue) and a C-terminal capping ankyrin (cyan); side chains of the randomized residues are shown in red. (lower part) Ribbon representation of the selected MBP binding ankyrin repeat protein (colours as above) This binder was isolated from a library of N-terminal capping ankyrin repeat, three designed ankyrin repeat modules and a C-terminal capping ankyrin repeat. G, General structure of human retinol-binding protein, a prototypic lipocalin (Schlehuber and Skerra, 2005). Ribbon diagram of the crystal structure of RBP with the bound ligand retinol (magenta, ball and stick representation). The eight antiparallel strands of the conserved β-barrel structure are shown in blue with labels A to H, and the four loops, which are highly variable among the lipocalin family, are colored red and numbered. The typical α-helix that is attached to the central β-barrel in all lipocalins, the loops at the closed end the N- and C-terminal peptide segments are shown in grey. The three disulfide bonds of RBP are depicted in yellow. H, Schematic representation of full-sized antibodies, multidomain and single-domain antigen-binding fragments (Saerens et al., 2008). Classical IgG consists of two light chains and two heavy chains. The light chain is composed of one variable (VL) and one constant (CL) domain, whereas the heavy chain has one variable (VH) and three constant domains (CH1, CH2, and CH3). The smaller antigen-binding antibody fragments are shown, that is, Fab, scFv, and the single-domain antibody (sdAb) fragment, VH.
Common to all approaches of finding a suitable scaffold are the following steps (Nygren and Uhlen, 1997; Smith 1998; Binz and Pluckthun, 2005):
• Choosing a small protein (domain) that is well expressed in bacteria or yeast and has good biophysical properties (stability, solubility)
• Creating a library of protein mutants by introducing diversity at a contiguous patch of the surface of the protein (e.g. loops)
• Using a genotype-phenotype display system (e.g. phage display) to select a range of binders to a therapeutic target of interest
• Screening the binders obtained by the selection procedure for the desired biological activity, e.g. by enzyme-linked immunosorbent assay (ELISA).
Mutations introduced in the protein scaffold to produce diversity may compromise the three-dimensional structure, stability and solubility of the protein scaffold, thus making the isolation of protein binders based on folding frameworks other than the immunoglobulin fold difficult. Nevertheless, more than 50 scaffolds have been described for the generation of new protein binders (reviewed in Binz et al., 2005; Gebauer and Skerra, 2009). Being mainly a topic of academic interest at first, nowadays the development and use of non-antibody classes of proteins is being pursued by small- and medium-sized biotechnology companies (Hey et al., 2005). The common basis for the successful development of well-designed protein drugs lies in the availability of both a well-behaving protein scaffold and an efficient selection or screening technology in order to find suitable candidate proteins in the large repertoire created by mutagenesis. The next sections will introduce selected classes of protein scaffolds (see also Figure 13.1) that have been successfully used and commercialized by small- and medium-sized companies. The first scaffold derived drug has been recently approved by the FDA for treatment of hereditary angioedema (see kunitz type domains), others are being investigated in clinical trials. Therefore, we will learn more in the near future about this interesting field of antibody alternatives.
Kunitz type domains
Proteases have important biological functions, they have been identified as key regulators of cellular processes such as ovulation, fertilization, wound healing, angiogenesis, apoptosis, peptide hormone release, coagulation and complement activation (Nixon and Wood, 2006). Because unregulated proteolysis in any of these processes will have undesirable effects, effective control of protease activity is critical. Indeed, unregulated activity has been implicated in the pathogenesis of many diseases (e.g. cancer, inflammatory diseases, chronic obstructive pulmonary disease (COPD), chronic pancreatitis, muscular dystrophy and Alzheimer’s disease (Naruse et al., 1999; Egeblad and Werb, 2002; Nunan and Small, 2002; Shapiro, 2002; Laval and Bushby, 2004; Parks et al., 2004), making proteases important target proteins. It is notable, that there are only a few marketed drugs (such as HIV protease inhibitors, angiotensin-converting enzyme inhibitors, the proteasome inhibitor bortezomib and the renin inhibitor aliskiren), since a key challenge in the discovery of new inhibitors is the ability to identify drugs which are both potent and specific. Structural similarities within the active site of most proteolytic enzyme families often result in a simultaneous inhibition of several family members, which can lead to an unacceptable toxicity profile. One solution to this problem represents the use of endogenous, engineered protease inhibitors that make more contacts with the target protease and therefore allow for tighter control over specificity. One type of endogenous inhibitors include the kunitz domain inhibitors, e.g. bovine pancreatic trypsin inhibitor (BPTI) (Ascenzi et al., 2003).
In 1992, Ladner and co-workers produced a small library of mutants of kunitz domain BPTI, displayed on phage and screened for the ability to bind to elastase (Roberts et al., 1992), a serine protease that is believed to play a causative role in a variety of lung diseases, including COPD and cystic fibrosis (Hautamaki et al., 1997). Isolates that were able to bind to elastase were expressed, purified and tested for inhibition of elastase. The P-loop (see Figure 13.1A), the region that was varied in the library, of the best inhibitor (Ki ∼1 pM) was transferred to a human kunitz domain without subsequent loss of potency (Ley et al., 1997). The resulting engineered molecule, depelestat (Dyax/Debiopharm SA), inhibited human neutrophile elastase (HNE) activity in the sputum of cystic fibrosis patients, was demonstrated to be safe in an aerolized format in monkeys and no adverse effects were reported in Phase I clinical trials (Delacourt et al., 2002; Grimbert et al., 2003; Nixon and Wood, 2006). Pharmacodynamic reports of a Phase IIa clinical trial have been shown to inhibit completely sputum HNE in 52.6% of the patients and to decrease interleukin-8 levels, a biomarker of inflammation, in the sputum of treated patients (Saudubray et al., 2003).
In 1996, another human kunitz domain, the lipoprotein-associated coagulation inhibitor (LACI-D1), was used as a scaffold for the successful isolation of a very specific and potent inhibitor of human kallikrein (Markland et al., 1996). Kallikrein is believed to be an important mediator of hereditary angioedema (HAE), a rare disorder with attacks of oedema in the hands, face, feet, abdomen and/or throat. This condition is caused by a genetic deficiency of C1 esterase inhibitor (C1-Inh), which is an endogenous inhibitor of plasma kallikrein. In Europe, HAE is treated with plasma-derived C1-Inh which attenuates attacks and may prove life-saving, but C1-Inh is expensive and requires the use of pooled blood product (Nixon and Wood, 2006). After an iterative selection and screening approach chosen by the authors (Markland et al., 1996), the plasma kallikrein inhibitor DX-88 (Dyax Corp/Genzyme Corp) was isolated and proved to be a potent inhibitor (Ki = 40 pM) with high selectivity (Table 1 in Williams and Baird, 2003). The use of DX-88 in a C1-Inh deficient mouse model demonstrated that DX-88 is effective in preventing changes in vascular permeability (Han et al., 2002). In December 2009, DX-88 (ecallantide) was approved by FDA for the treatment of HAE.
Tenth type III domain of fibronectin (Adnectins)
The tenth type III domain of human fibronectin (10Fn3) is a 94 aminoacid residue structural protein with an immunoglobulin-like fold that is used as an antibody-mimic scaffold. High thermal stability (TM = 90°C) and solubility (>15 mg/mL), high expression yields in Escherichia coli and the lack of cysteines in its structure are attractive properties that make the 10Fn3 a good scaffold candidate (Xu et al., 2002). Three of the solvent exposed loops in 10Fn3, named BC, DE and FG are structurally analogous to the VH complementary-determining regions (CDR) (Figure 13.1B).
The first report on the use of the 10Fn3 domain as a protein with artificial binding sites was published in 1998 (Koide and Koide, 1998). In that study, a library of 108 distinct 10Fn3 mutants was made by deleting three residues in the FG loop and by randomizing five residues in the BC loop and four residues in the FG loop. The library, displayed on phage, was used in affinity selection experiments with immobilised ubiquitin as model target protein. After five rounds of panning, clones were randomly picked for sequencing. A clone designated as Ubi4-Fn3 dominated the population of selected protein mutants and was subjected to further analysis. Ubi4-Fn3 was demonstrated to bind in phage enzyme-linked immunosorbent assay (ELISA) experiments. However, when expressed as a single domain protein in E. coli, Ubi4-Fn3 exhibited low solubility at neutral pH, unspecific binding to the dextran matrices of the size-exclusion chromatography column and the dextran coated biosensor chips used for the subsequent characterization of Ubi-Fn3. More importantly, the affinity was relatively low; the IC50 determined by competition ELISA was 5 µM.
In order to select 10Fn3 domains binding to a target of interest with improved properties than Ubi4-Fn3, an alternative library design in combination with a fully in vitro selection system (mRNA display (Wilson et al., 2001)) was used for the isolation of 10Fn3 variants binding to tumour necrosis factor-α (TNF-α) (Xu et al., 2002). Clones from the ninth and tenth round of selection were cloned in E. coli, sequenced and expressed. Affinity constants were determined by incubating the in vitro translated 35S-methionine labelled proteins with biotinylated TNF-α at different concentrations. The solution containing the 10Fn3 mutants bound to TNF-α were aspirated by vacuum onto a membrane coated with streptavidin, therefore capturing protein complexes on the membrane. Binding was analysed by measuring the radioactivity on the membrane, the KD for selected mutants were found in the range of 1-24 nM. Further affinity maturation procedures revealed KD values around 100 pM, the best being 20 pM, and analytical gel filtration showed that the apparent molecular weight of purified, soluble wild-type (wt) and mutant domains was consistent with the variants being monomeric. The reported affinities, especially for TNF-α, are very high. However, these values should be evaluated critically because first, the capture step of the biotinylated TNF-α-Fn3 protein complexes was performed in a solid phase format, and second, TNF-α is homotrimeric protein, so avidity effects may contribute to the high apparent affinity observed.
Other 10Fn3 variants were shown to recognize ανβ3-integrin expressed on cell surface and inhibiting ανβ- dependent cell adhesion (Richards et al., 2003) and to bind to vascular endothelial growth factor receptor 2 (VEGF-R2) inhibiting the activation by VEGF-A, -C and –D (Parker et al., 2005; Getmanova et al., 2006). Indeed, the test case of 10Fn3 for the in vivo behaviour in human patients is the VEGF-R2 binding CT-322 (KD = 11 nM), currently in clinical development by Bristol Myers Squibb in cancer patients (Choy et al., 2002; Molckovsky and Siu, 2008). The CT-322 10Fn3 domain is coupled via a C-terminal cysteine to a 40-kDa polyethylene glycol (PEG) to increase the size of the molecule above the threshold for kidney-mediated clearance. In a Phase I study in patients with solid tumours, CT-322 administered intravenously at 1 mg/kg showed a terminal half-life of 68.7 hours, based on the first dose of a weekly dosing regimen (Molckovsky and Siu, 2008). The half-life in humans is substantially shorter than that of IgG (7–26 days), as expected for a molecule lacking FcRn binding, and also shorter than the 14-day half-life reported for Cimzia, a pegylated Fab that binds to TNF-α (Choy et al., 2002). Still, the pharmacokinetics of CT-322 supported weekly intravenous dosing, and a biomarker for VEGF-R2 blockade showed sustained elevation over several repeated weekly doses. Immunogenicity was also assessed in this Phase I trial showing that 31 out of 39 patients developed antibodies to CT-322 (Beyond Antibodies Conference, 2009, San Diego) but these did not lead to a clinical significant immunogenicity (Bloom and Calabro, 2009). As for other biologics, immunogenicity should be monitored carefully for this scaffold, especially for the generation of cross-reactive antibodies binding not only to the injected mutant, but also to the endogenous fibronectin domain. In the near future we will learn more about this scaffold, since patients are currently being enrolled in a Phase II trial in glioblastoma multiforme, in which CT-322 will be tested alone or in combination with irinotecan (Campto, Pfizer), a cytostatic agent inhibiting DNA-topoisomerase I. The technology of using the 10Fn3 domain was commercialized by Adnexus, which was acquired by Bristol Myers Squibb for $430 million in 2007, proving the interest and significant potential of antibody alternatives in the field of pharmaceutical biotechnology.
A-domains (Avimers)
A family of A-domains (Huang et al., 1999; North and Blacklow, 1999) has been described to be suitable as a scaffold for the generation of new binding proteins (Silverman et al., 2005). A-domains occur as strings of multiple domains in several cell-surface receptors (Figure 13.1C). Domains of this family bind over 100 different known targets, including small molecules, proteins and viruses (Krieger and Herz, 1994; Gliemann, 1998). Such a target is typically contacted by multiple A-domains with each domain binding independently to a unique epitope, thereby binding with high avidity. Each of the 217 human A-domains comprises approximately 35 amino acids and the domains are separated by linkers that average five amino acids in length. Native A-domains fold quickly and efficiently to a uniform, stable structure mediated by calcium binding and disulfide formation. A conserved scaffold motif of only 12 amino acids is required for this common structure (Koduri and Blacklow, 2001). Stemmer and co-workers have designed a phage display library of A-domains, consisting of a conserved consensus sequence in the scaffold motif and variable amino acids at different positions (Silverman et al., 2005). Like other directed evolution technologies, the process the authors developed to isolate binding proteins, included multiple recursive cycles, each consisting of library generation and screening in functional assays. However, rather than mutagenizing the protein between cycles (e.g. for affinity maturation purposes), they added a new domain library adjacent to the domain(s) selected in previous rounds.
The result of this process is a single protein chain containing multiple domains, each of which having a separate binding function. The A-domains are therefore called ‘avimers’, from avidity multimers. A heterotetramer consisting of three IL-6 binding A-domains and an IgG binding domain (named C326) showed a remarkable affinity (picomolar range) to its target and exhibited sub-picomolar IC50s in cell-proliferation assays (Silverman et al., 2005). Moreover, C326 completely abrogated acute-phase protein induction by human IL-6 in mice in a dose-dependent manner, suggesting that C326 inhibited IL-6 functions in vivo. The technology of using the modular platform of A-domains was commercialized by Avidia Inc. that was acquired by Amgen for $290 million in 2006. A placebo-controlled Phase I study of C326 was registered in the United States in September 2006, with patients suffering from Crohn’s disease. Since then, to our knowledge, new results have not been published in the public domain. Recent data (Beyond Antibodies Conference, 2009, San Diego) indicate that Amgen is further engineering avimers for the neutralization of IL-6 mediated effects.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


