Acids and bases can be classified as strong through to extremely weak, based on their pKa (Table 6.3). When strong acids react with strong bases the reaction tends to completion, as both species will be fully ionised, a process termed neutralisation. For example,
(6.1)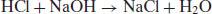
Table 6.3 Descriptions of acid and base strength (data from Stahl and Wermuth (2011)).
| pKa | ||
| Description | Acid | Base |
| Very strong | <0 | >14 |
| Strong | 0–4.5 | 9.5–14 |
| Weak | 4.5–9.5 | 4.5–9.5 |
| Very weak | 9.5–14 | 0–4.5 |
| Extremely weak | >14 | <0 |
In this instance the salt formed will precipitate, once it is present at a concentration beyond its solubility at any particular pH. Most drug substances, however, are either weak acids or bases, in which case acidic and basic character is usually based on the Brønsted–Lowry definition. This posits that an acidic compound is a proton donor and a basic compound is a proton acceptor. The removal of a proton from an acid produces a conjugate base (A−) and addition of a proton to an acceptor produces a conjugate acid (BH+):
(6.2)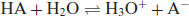
(6.3)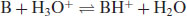
Note that the Brønsted–Lowry definition requires acidic species to have an ionisable proton but does not require basic compounds to possess a hydroxide group, simply that they can accept a proton (the theory does not consider KOH to be a base, for instance, but a salt containing the basic OH− moiety). Note also that protons do not exist in aqueous solution in isolation but as the hydronium ion. In the case of a weak base (B) reacting with a strong acid, the conjugate acid and conjugate base may then form a salt:
(6.4)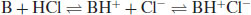
When a salt dissolves in water it will dissociate (in the majority of cases, with exceptions including some mercury salts). Assuming dissolution of a basic salt then the species in solution is the conjugate acid. The conjugate acid can donate its proton to water, reforming the free base:
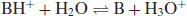
All of the reasons for the change in solubility of salts are encompassed in Equation (6.5). A basic salt contains the conjugate acid of the drug substance. Upon dissolution the conjugate acid donates its proton to water and the free base is formed. The solute is thus the free base, but the pH of the solution in which it is dissolved has reduced because of the donated proton. Recall from Chapter 4 that the solubility of weak bases increases as the pH of the solution reduces. Thus dissolution of a basic salt increases solubility over the free form because there is a concomitant reduction in pH of the solution.
Since the pH of a solution of a dissolved acid is given by
(6.6)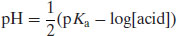
It follows that because the acid species is BH+ then
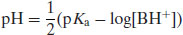
Example 6.1 What is the pH of a 0.2M solution of ergotamine tartrate (pKa 6.25)?
From Equation (6.7),
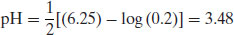
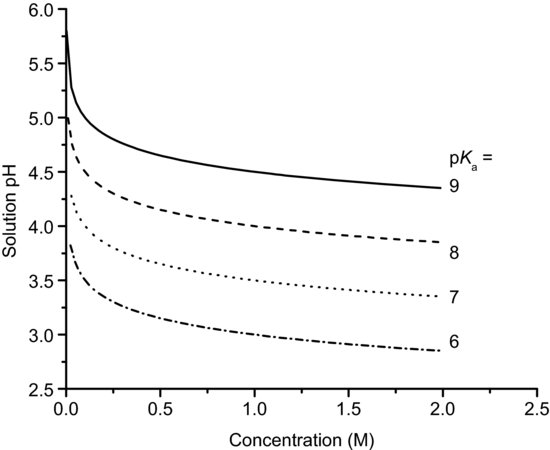
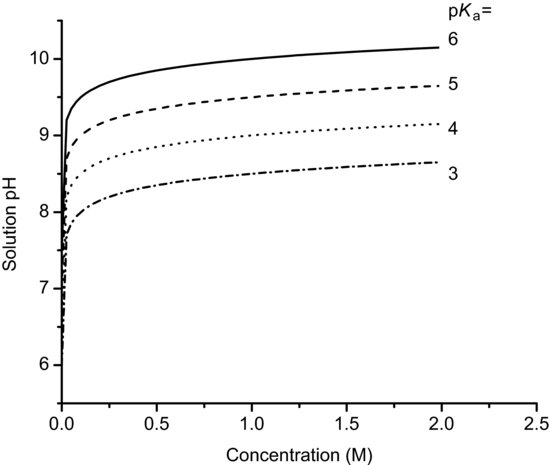
Figure 6.1 shows the solution pH as a function of concentration for basic salts over the pKa range 9–6, calculated with Equation (6.7). It is apparent that even a small concentration of salt leads to a significant change in solution pH.
A similar situation occurs for the reaction of a weak acid with a strong base:
(6.8)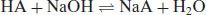
Upon dissolution of an acidic salt the conjugate base is formed:
(6.9)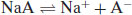
The conjugate base can then accept a proton from a hydronium ion, reforming the free acid and increasing the pH of the solution:
(6.10)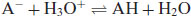
The solute is thus the free acid, but the pH of the solution in which it is dissolved has increased because a hydronium ion has been removed from the system. Recall again from Chapter 4 that the solubility of weak acids increases as the pH of the solution increases.
The pH of a solution of base is given by
(6.11)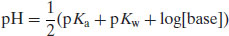
Since A− is a conjugate base then
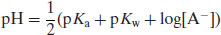
where pKw is the self-ionisation constant of water, usually assigned a value of 14, although the value shows considerable temperature dependence (Table 6.4). This is why neutral pH is 7.0 only at 24 °C and ranges from 7.5 at the freezing temperature of water to a little over 6.0 at the boiling temperature of water. Figure 6.2 shows the solution pH as a function of concentration for acidic salts over the pKa range 3–6, calculated with Equation (6.12).
Table 6.4 Values for the self-ionisation constant of water as a function of temperature (data from Bandura and Lvov (2005)).

Example 6.2 What is the pH of a 0.1 M solution of diclofenac sodium (pKa 4.0)?
From Equation (6.12),
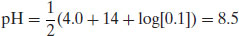
Several consequences arise from this discussion. One is that salt formation might not be best achieved in an aqueous solution, since dissolution of a salt in water generally results in formation of the free acid or base. For this reason salts are often formed in organic solvents (although it has been suggested that the addition of at least 10% v/v water to crystallisation solvents can improve salt formation for poorly water soluble bases; Tarsa et al., 2010). Secondly, the increase in solubility of a salt over the corresponding free acid or base is a result only of the change in pH upon dissolution. The intrinsic solubility of the free acid or base does not change.
Study question 6.1 What does this mean for solubility of salts in buffered media?
- Salts form when acids react with bases. Weak acids are reacted with strong bases and vice versa.
- Acidic drugs are proton donors, basic drugs are proton acceptors.
- Forming a salt does not change the pKa of the parent molecule.
- The increase in solubility of a salt is because of the change in pH upon dissolution. Dissolution of an acidic salt increases pH and dissolution of a basic salt decreases pH.
6.2.1 Selection of a salt-forming acid or base
In order for the salt to form there must be a sufficient difference in pKa values between the acid and base (the reactivity potential). For the transfer of a proton from an acid to a weak base the pKa of the acid must be less than that of the weak base and vice versa. As a general rule, a difference in pKa (ΔpKa) of 3 is indicated, although salt formation can occur with smaller differences (for instance, doxylamine succinate forms spontaneously even though ΔpKa is only 0.2; Wells, 19881). The reason for this is to ensure that both species are ionised in solution, thus increasing the chance of interaction. Childs, Stahly and Park (2007) note that if ΔpKa is greater than 3 then salts will invariably form while if ΔpKa lies between 3 and 0 then knowledge of ΔpKa per se is not predictive of whether salt formation will occur. In cases where ΔpKa is less than zero co-crystal (Chapter 7) formation is the more likely outcome (Bhogala, Sasavoju and Nangia, 2005).
Brittain (2007, 2008) points out that if one considers the formation of the salt of a weak acid (such as ibuprofen) with a strong base,
(6.13)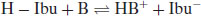
Then the position of equilibrium is given by
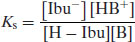
and so
(6.15)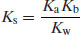
or
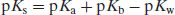
Expressed in terms of pKa only, Equation (6.16) becomes
(6.17)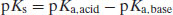
Thus, with knowledge of the pKa values of the drug substance and salt-forming acid or base (as appropriate) it is possible to calculate the position of equilibrium of the salt formation reaction. Assuming that equimolar concentrations of acid and base are reacted initially (C) and that upon attainment of equilibrium some fraction of salt (x) is formed, then Equation (6.14) becomes
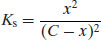
If C is set equal to 1, so fractions are calculated, rearranging Equation (6.18) leads to the quadratic form:
(6.19)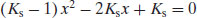
which can be solved for x:
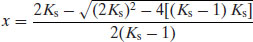
Equation (6.20) allows determination of the fraction of salt formed as a function of ΔpKa between the acid and base. The data in Figure 6.3, generated with Equation (6.20), show the fraction of salt formed (as a percentage) versus the acid pKa for a basic drug substance of pKa 7.5. The general rule noted above that ΔpKa should be greater than 3 is explained by this relationship, because when ΔpKa = 3 nearly 97% of the reactants will form a salt (and if ΔpKa >4 more than 99% of the reactants form a salt). Brittain (2008) shows that the same relationship is true for an acidic drug. Thus, selection of a salt former starts with knowledge of its pKa and the pKa of the drug substance. The pKa values of some of the most common salt-forming acids and bases are given in Tables 6.5 and 6.6 (note that these values are determined in water and thus will change if organic solvents are used, which is likely). The top 10 anions and cations by frequency for drugs in the 2006 USP are shown in Table 6.7.
Figure 6.3 Percentage of salt formed as a function of acid pKa when reacted with a basic drug of pKa 7.5.
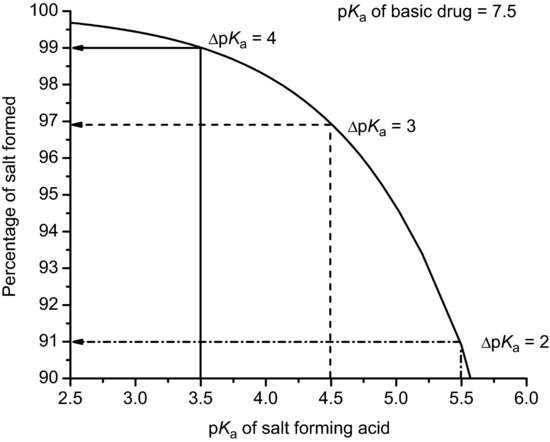
Table 6.5 Values of pKa for selected pharmaceutical acids (data from Stahl and Wermuth (2011)).

Table 6.6 Values of pKa for selected pharmaceutical bases (data from Stahl and Wermuth (2011)).
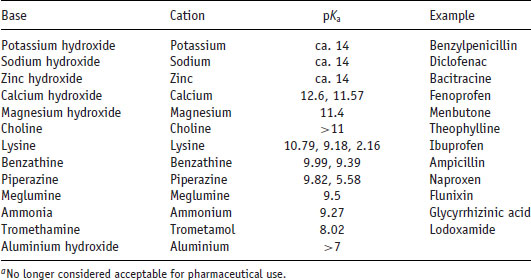
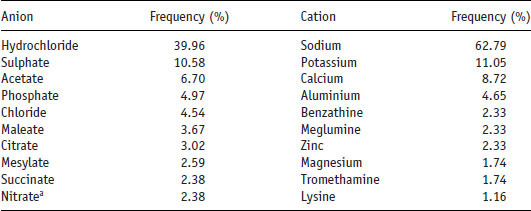
Table 6.7 Frequency of pharmaceutical anions and cations of drugs in USP 29-NF24 (data from Kumar et al. (2008)).
Study question 6.2 Why do you think the hydrochloride salt is the most common form for basic drug substances? What disadvantages might using this salt have?
Stahl and Wermuth (2011) organise salt formers into three categories, which may be used as a guide to selection:
- First class salt formers are those that form physiologically ubiquitous ions or that occur as metabolites in biochemical pathways (including the hydrochloride and sodium salts). As such, they are considered to be unrestricted in their use.
- Second class salt formers are those that are not naturally occurring but that have found common application and have not shown significant toxicological or tolerability issues (such as the sulphonic acids).
- Third class salt formers are those that are used in special circumstances to solve a particular problem. They are not naturally occurring nor in common use.
An additional factor to consider is that the salt formed should exist as a crystalline solid, to enable ease of isolation and purification. Amorphous salts are highly likely to cause problems in development and use and so should be avoided.
6.2.2 Salt screening
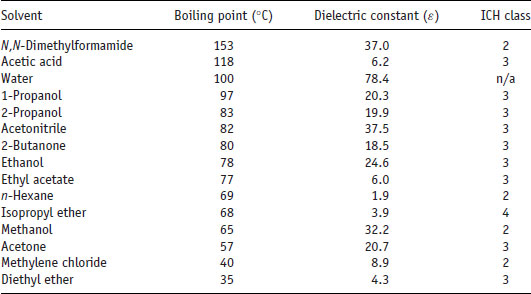
Once potential salt formers have been selected they must be combined with the drug substance in order to see which preferentially form salts. Since the potential number of permutations and combinations of salt formers and solvents is large, a convenient method for salt screening is to use a microwell plate approach. A small amount of drug substance (ca. 0.5 mg) in solvent is dispensed into each well of a 96-well plate. To each well is added a solution of potential counterion. It is possible to construct the experiment so the effect of the solvent is examined in the x dimension and the effect of the counterion is examined in the y dimension. Solvents should be selected carefully and those listed as class I by ICH Guideline Q3C(R5) (2011) (i.e. those that are either known or suspected carcinogens or that pose an environmental danger) should be avoided. Typically used solvents are listed in Table 6.8, with their ICH class.
Table 6.8 Properties of some common solvents used for salt screening (data from Huang and Tong (2004)).
After an appropriate length of time, the presence in each well of salt crystals is checked with an optical device (for instance, a microscope or a nephelometer). If no crystals are seen then the plate can be stored at a lower temperature. If the reduction in temperature does not cause precipitation then as a last attempt the temperature can be increased to evaporate the solvent (although care must be taken in this case during subsequent analysis because the isolate may contain a simple mixture of unreacted drug and salt former, rather than the salt itself).
Once a potential salt has been identified preparation can be undertaken with slightly larger sample masses (10–50 mg). X-ray powder diffraction (XRPD) may be used to get a preliminary idea of polymorphic form while melting points may be determined with a melting point apparatus, hot-stage microscopy (HSM) or differential scanning calorimetry (DSC). Examination with HSM, if operated under cross-polarised filters, allows quick visual confirmation of crystallinity and melting and any other changes in physical form during heating, while analysis with DSC provides the enthalpy of fusion in addition to the melting temperature (and so access to ideal solubility). Additional analyses with thermogravimetry analysis (TGA) and dynamic vapour sorption (DVS) will provide information on residual water content, the presence of the salt in a solvated or hydrated state and/or hygroscopicity tendency. All of these experiments can be performed with just ca. 50 mg of salt, if available.
Of course a salt screen such as this may result in a large number of potential salt candidates and so an approach is needed to select a drug substance for development. Morris et al. (1994) published an integrated approach to salt candidate selection which is effectively a decision tree method based on easily measured physicochemical properties (Figure 6.4). Following isolation of crystalline salts the hygroscopicity of each is determined (since this is easy to measure, does not in principle destroy the sample and gives a likely indication of stability and processability). Those salts with acceptable properties are advanced to tier 2, where physical form stability with respect to relative humidity (RH) and solubility are measured. This would indicate the likelihood that the salt would change polymorphic form during processing or storage and allows selection of a salt with satisfactory solubility (bearing in mind that the salt with the highest solubility would not necessarily be the easiest to manufacture or formulate). Finally tier 3 testing involves thermal and photostability assessment under accelerated stress conditions. At each tier the time and cost associated with testing increases, so it is good practice to reduce the number of candidates at each stage.
Figure 6.4 A decision tree for salt selection based on characterisation of physicochemical properties (data from Morris et al. (1994)).
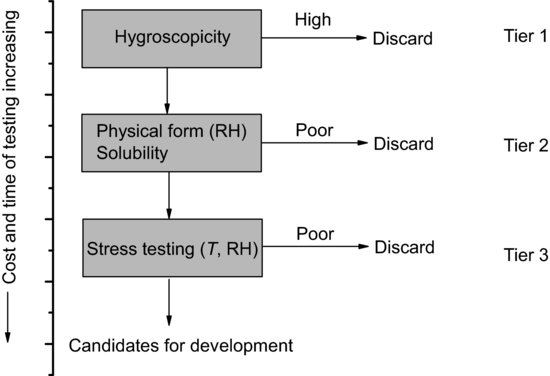
Of course, such a regimented scheme must be undertaken with an open mind. The calcium and sodium salts of fenoprofen have equivalent bioavailability, distribution and elimination profiles, but the calcium salt is less hygroscopic so it is the marketed form (Lemke et al., 2008). Ranitidine hydrochloride, conversely, is extremely hygroscopic (Teraoka, Otsuka and Matsuda, 1993) yet is a successful product. Similarly, sertraline hydrochloride has around 28 reported polymorphic forms (Remenar et al., 2003). Both of the latter drugs would have been discarded according to the scheme in Figure 6.4.



