New Terminology212
Safety Versus Toxicity212
Safety Pharmacology214
Early Safety Tests219
Hepatic Toxicity223
Summary225
The main aim of safety pharmacology is to define the optimal way in which potentially valuable drugs can be utilized without causing harm. An important difference between testing for primary activity and safety is that, in the former, what to look for is defined; in safety testing it is not known what should be looked for. Under these circumstances, a great many tests must be undertaken to produce confidence that harmful effects will not be seen. This chapter discusses the breadth of assays and testing procedures available to assess the safety of a new drug entity and their use in determining potentially serious toxicity in molecules at a very early stage. In addition, the important difference between toxicity due to elevated drug exposure (intrinsic toxicity) and that due to stochastic opportunity (idiosyncratic toxicity) is considered. This latter toxicity is difficult to predict in drug testing and thus poses a serious risk in the drug development process, since even with exhaustive testing, once a drug enters the greater population, very rare negative events (the probability of which may be infinitesmal in drug development testing) may still be found. Some common in vitro simple tests to quickly detect chemical scaffolds that interact with autonomic receptors, block the hERG channel, are cytotoxic or are mutagenic are discussed. In addition, hepatotoxicity, a very common toxic drug effect due to the fact that high concentrations of drugs reach the liver (and the fact that the liver is designed to chemically interact with drugs) is considered.
Keywords
carcinogenesis, cytoxicity, idiosyncratic toxicity, intrinsic toxicity, mutagenesis.
Introduction
In addition to having primary activity and being able to enter the body, access the appropriate tissue and have sufficient target presence to achieve therapeutic utility, a drug must cause no harm to the host. Therefore, the third important structure–activity relationship that must be explored for a drug is safety pharmacology. The human body is a finely tuned symphony of biochemical reactions and physiological functions, and miscues can cause the system to go awry. It is unrealistic to suppose that extreme amounts of almost any substance won’t eventually cause harm; as put by Paracelsus (1493–1541):
“… all things are poison and nothing is without poison. Solely the dose determines that a thing is not poison …”
The concept of “safety” can also be relative. As pointed out in the discussion of target validation in Chapter 1, one of the criteria in the choice of a favorable target for an antagonist is that the knockout mouse (genetically altered such that the target is not expressed in tissues) is healthy, i.e., the organism can do without the target. Thus it was observed that CCR5 knockout mice (lacking the CCR5 receptor) were healthy, thereby indicating that the CCR5 receptor is redundant and not required for life. While this spurred on pursuit of CCR5 as a target for HIV-1-mediated AIDS infection, the idea that CCR5 is redundant and not required for health was later shown to be simplistic. In this case, there is a human counterpart to the CCR5 knockout mouse, namely a population of people possessing a Δ32 CCR5 deletion in the receptor that causes it not to be expressed on the cell surface; in essence, human equivalent “knockouts.” These people appeared to have normal health; however, as data accumulated this concept of “healthy” began to be questioned. Specifically, it had been shown that Δ32 subjects have a higher than average incidence of health abnormalities (i.e., greater incidence of liver disease and schlerosing cholangitis, [1] risk of death in Nile Virus disease, [2] greater mortality after liver transplantation, [3] mild immunodeficiency[4]). The point of these data for this chapter is to suggest that chemical intervention into any physiological function almost necessarily brings with it a risk of imbalance and consequent risk of harm. Therefore, it is a defensible statement to suggest that all drugs, if given in sufficient dosage, may have harmful side-effects and pharmacological safety is simply a matter of relative benefit to risk of harm. This chapter will consider this benefit-to-risk ratio for drugs.
New Terminology
The following new terms will be introduced in this chapter:
• Carcinogenesis: The creation of cancer where normal cells are transformed into cancer cells
• Cytotoxicity: The quality of being toxic to cells to detrimentally affect cell metabolism, function, growth and to subsequently induce damage.
• Idiosyncratic toxicity: Toxic effects due to stochastic probability occurring when a combination of favorable conditions coalesce.
• Intrinsic toxicity: Toxicity due to elevation of dosage above a threshold for toxicity, i.e., it is observed whenever this dosage is exceeded.
• MTD: Maximum tolerated dose – applies to long-term studies and refers to the largest dose that causes no obvious signs of ill health.
• Mutagenesis: Induction of genetic change in a cell through alteration of cell genetic material (usually DNA).
• NOAEL: No observed adverse effect level – the largest dose causing no observed toxicity or undesirable physiological effect.
• NOEL: No observed effect level – the threshold for producing a pharmacologic or toxic effect.
• NTEL: No toxic effect level – the largest dose in most sensitive species that produces no toxic effect.
Safety Versus Toxicity
Drug safety is often discussed in terms of “toxicology” when it is really more appropriate to focus on the term safety, defined as “the condition of being safe from undergoing … hurt, injury.” Presupposing that a drug will cause harm if used inappropriately or in too high a dose, the aim is to define the conditions whereby a drug can be used effectively to heal with minimal risk of toxicity (defined as “containing or being a poisonous material … capable of causing death or serious debillitation”). For example, the drug theophylline is used intravenously in emergency rooms for acute bronchoconstriction in children. While a serum concentration of 15μg/mL produces life-saving bronchodilation, 25μg/mL causes mild side-effects which become potentially serious at 35μg/mL and severe at 42μg/mL. Thus, less than a three-fold increase in serum plasma levels can lead to toxicity. The key to theophylline’s value is the fact that it is used in a strictly controlled setting (intravenous administration under a physician’s supervision with constant surveillance). In this case, the “safety” of theophylline is an extremely qualified parameter.
There are categories of toxicity based on mechanism:
• Undesired but expected effects: These result from the primary pharmacology of the drug occurring by the therapeutic mechanism but in tissues other than the primary therapeutic organ. These usually cannot be avoided. Examples of this type of toxicity are digital tremor with β2-adrenoceptor bronchodilators. Since β2-adrenoceptors mediate bronchodilation and unwanted response (digital tremor), any such agonist will produce both effects. Restricted route of administration (aerosol) is used to minimize these side-effects.
• Desired excessive effects: These result from the primary pharmacology of the drug acting on the therapeutic organ. An example of this is insulin-induced hypoglycemic reaction. These also cannot be avoided and come with excessive dose.
• Undesired unexpected effects: These occur by mechanisms different from the primary therapeutic mechanism of action of the drug. An example of this is the dry mouth coming from antimuscarinic effects of antihistamines such as diphenhydramine.
• Poorly predictable effects: These are the most problematic in that they are difficult to detect. They consist of drug allergies, idiosyncratic effects, mutagenesis, carcinogenesis and drug dependency.
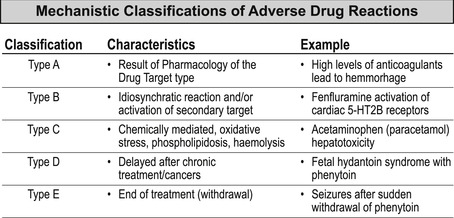
There are other classifications of toxicity used such as the Type A to E listing of adverse drug reactions (see Box 10.1).
Box 10.1
An adverse drug reaction (event) (ADR) can be defined as any undesired, noxious or unintended event occurring from a normal dosage (used for prophyllaxis, diagnosis or treatment) of the drug. The Federal Drug Administration regards any outcome leading to death, risk of death, hospitalization, disability or required intervention to prevent permanent impairment or damage as adverse; these account for approximately 5% of all hospital admissions per year. Treatment failure, intentional or accidental overdose are not considered ADRs. Below is one classification of ADRs based on drug mechanism of action (data from [13]).
 |
Safety Pharmacology
There is a basic difference between safety assessment and determining primary drug efficacy. In the latter, researchers know what to look for; in safety assessment they do not. A hazard cannot be characterized until it is identified. Therefore, safety pharmacology involves the dual tasks of hazard identification and risk assessment. Hazard identification basically examines the profile of a drug and analyzes the potential harmful effects that can, and do, occur. Risk assessment then quantifies the probability that they will occur during clinical or accidental exposure. From this point other analyses are done to yield possible dose–response relationships for these effects. This involves issues such as reversibility and determination of NOAEL (no observed adverse effect level; see Chapter 8) and parameters for clinical monitoring. Other features of these analyses include examination of physical risk factors (age, gender, susceptible patient populations), environmental risk factors (conditions present that may enhance toxicity/pharmacological interactions) and genetic risk factors (genetic determinant for susceptibility to toxicity, known gene polymorphisms).
Safety assessment of drugs is an extremely important endeavor in that if a hazard is not identified, patients may be harmed. Moreover, in the drug development process, resources increase exponentially as candidate molecules reach the end of the development process; to reduce expenditure of large resources, candidates that will not be successful due to toxicity must be identified as soon as possible (see Box 10.2). As safety testing begins, a number of lines of investigation are initiated (see Table 10.1):
Box 10.2
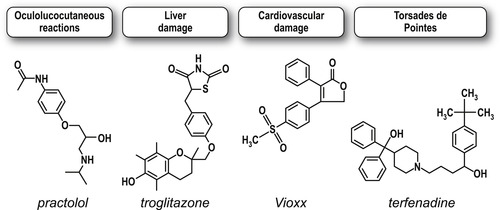
A prospective drug has incurred nearly 90% of its cost of discovery and development by the time it is in Phase III testing. Therefore it is of paramount importance that an unsuitable molecule be eliminated from development before costs rise to this level. However, even more devastating is the discovery of serious toxicity after a drug has been approved and is on the market. Below are four examples of drugs that demonstrated rare but extremely serious toxicity after marketing, causing them to be withdrawn from the market.
 |
| Development Phase | Safety Activity |
|---|---|
| Lead to candidate | • Acute toxicology • hERG • Ames test/cytotoxicity • Early safety prediction • 2° receptors |
| Candidate selection to first human dose | • Genotoxicity • Safety pharmacology dose ranging • 14–28 day toxicology studies |
| Patient phase 2a/2b studies | • 3/6/9/12 month toxicology studies • Reproductive toxicology studies • Immunotoxicology studies |
| Phase 3 clinical studies | • Carcinogenesis • Reproductive toxicology |
• Safety pharmacology: Detection of undesirable pharmacodynamic effects on specific organ systems. Some of these tests are simple in vitro experiments that can be done very early on in the development process much like in vitro ADME studies (CaCo-2, hepatic enzymes; vide infra).
• Genetic toxicity: Examination of possible gene mutation and chromosomal damage.
• Single/repeat dosing studies: Detailed examination of target toxicity and local tolerance.
• Reproductive toxicity: Effects on embryo-fetal development, fertility, parturition, post-natal development.
• Carcinogenicity: Risk of development of tumors.
• Special studies: Immunotoxicology, phototoxicology, environmental health and safety.
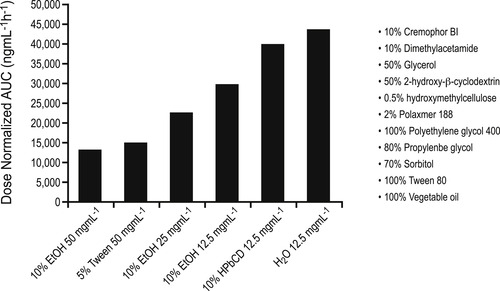
A major tool in these endeavors is the repeat dosing study. In this procedure the goal is to develop an understanding between exposure to drug and observed untoward effects, and also to assess the potential for reversibility. Response can be directly observed or the study may seek to find biomarkers for effects. The overall aim is to assess organ toxicity and safety associated with repeat dosing to predict risk in humans; the object is to specifically define the NOEL (no observed effect level), MTD (maximal tolerated dose) and NOAEL (no observed adverse effect level). The first step in this process is to achieve high exposures of systems to the drug in vivo. For this, pharmacokinetic effects may need to be observed at doses extended far beyond what is required therapeutically, i.e., dosing must continue (and exposure be increased) until adverse effects are detected. [5] Therefore, cases of limited exposure due to solubility (see Chapter 8 for discussion of solubility limitation for exposure to the hepatoprotective agent YH439) can be problematic. In these cases extended exposure protocols may be required which may not be applicable to therapeutic dosing. Figure 10.1 shows the effects of some solvents used for administration of experimental candidate molecules to optimize solubility and ultimately, exposure.