Fig. 22.1
Tobacco carcinogen and inflammatory response-induced reactive oxygen species (ROS), bacterial, and viral products mediate inducible and genomic alterations in the PI3K-NF-κB, TP53/63, and KEAP1/NRF2 pathways in HNSCC. (a) Tobacco carcinogen and inflammatory cell-induced ROS cause DNA damage and degrade sensor and ubiquitin ligase KEAP1, inducing activation of the classical Inhibitor-κB kinase (IKK)-NF-κB pathway, which elicits transcription of cancer promoting genes. Carcinogen and ROS-induced genomic mutations in tumor suppressor TP53 and amplifications causing overexpression of oncogenic family member ΔNp63 and PI3-Kinase (PI3K) result in loss of growth control and enhance NF-κB signaling. Classical NF-κB activation may be enhanced by bacterial and viral products, inflammatory and growth factors. (b) Alternative NF-κB pathway activation (and TP53 inactivation) can be directly mediated by HPV E6 and EBV LMP1 oncogenes. ROS-induced KEAP1 and NRF2-mediated transcription of endogenous antioxidants may also be compromised by genomic alterations
22.3 Activation of NF-κB and Inflammation-Related Free Radical Damage
The NF-κB/REL family transcription factors are aberrantly activated in HNSCC and other cancers, and critically promote cell survival, inflammation, and angiogenesis (Fig. 22.1) (Van Waes 2007). As aforementioned, nicotine and tobacco metabolites can promote PI3K-Akt and PKA signaling, and my laboratory showed that PI3K and PKA contribute to aberrant transactivation of NF-κB observed in HNSCC (Fig. 22.1) (Bancroft et al. 2002; Arun et al. 2009). Additionally, many injury and pathogen inducible signal pathways converge to activate NF-κB (Van Waes 2007). Carcinogen and ROS-induced DNA damage can promote sumoylation and activation of Inhibitor-κB kinases (IKKs), which mediate NF-κB nuclear translocation and activation. Further, ROS can promote degradation of ubiquitin ligase KEAP1, enhancing IKK-mediated signaling (Fig. 22.1). Bacterial, human papilloma virus (HPV) and Epstein Barr Virus (EBV) pathogens have also been implicated in development of HNSCC, and can induce activation of Toll-Like Receptor, IKKs, and alternative pathways that promote NF-κB activation (Van Waes 2007; James et al. 2006; Szczepanski et al. 2004) (Fig. 22.1).
The consequences of such chronic injury-induced signal activation of NF-κB are pathologic. NF-κB promotes expression of Cyclin D1 and BCL–XL genes that promote cell proliferation and survival of HNSCC cells (Van Waes 2007; Lee et al. 2008; Duan et al. 2007). NF-κB also promotes expression of angiogenesis factors IL–6, IL–8, GRO1, and VEGF (Duffey et al. 1999; Bancroft et al. 2001; Loukinova et al. 2001) that recruit and activate monocytic and myeloid inflammatory cells (Loukinova et al. 2000; Young et al. 2001). Activated myeloid-derived cells produce ROS, which likely further exacerbates cell and DNA damage, related signaling and mutations, and compromises immune defenses to malignant cells (Kotsakis et al. 2012; Vasquez-Dunddel et al. 2013).
22.4 Role TP53 and p63/PI3KCA Genetic Alterations in Genomic Instability and Inflammation in HNSCC
Among genetic alterations, mutation or deletion of TP53 is the most frequent, occurring in over 70 % of 279 HNSCC tumors studied as part of The Cancer Genome Atlas (TCGA) (Fig. 22.2) (TCGA Network 2015). TP53 is a ROS and DNA damage inducible transcription factor that mediates growth arrest and DNA damage repair, or death of cells with irreversibly damaged DNA (Fig. 22.1). Hence, TP53 serves as the “Guardian of the genome,” and its loss leads to uncontrolled proliferation, genomic instability, and progressive genomic alterations (Lane 1992; Stiewe 2007). Among the gains, amplification of the locus containing the gene encoding a TP53 family oncogene ΔNp63, and amplification or activating mutations of PIK3CA, the PI3kinase catalytic subunit alpha, are prevalent (Fig. 22.2) (Walter et al. 2013). In ~20 % cases, ΔNp63 and PIK3CA are included in the same amplicon, while overall, PIK3CA is amplified or mutated in 36 % of cases. Interestingly, these genomic alterations in TP53, ΔNp63, and PIK3CA may contribute to inactivation of TP53-dependent responses, and constitutive activation of transcription factor Nuclear Factor-κB, cell survival, and host inflammatory responses (Fig. 22.1) (Yang et al. 2011; Vander Broek et al. 2014; Du et al. 2014; Cooks et al. 2013), that further enhance free radical production and cumulative DNA damage, resulting in cancer progression.
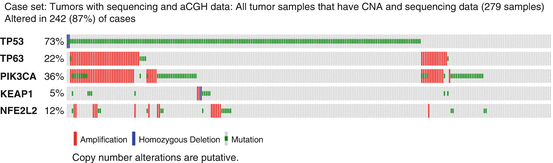
Fig. 22.2
Genomic alterations in TP53, TP63, PIK3CA, KEAP1, and NFE2L2 genes in HNSCC. Publically available data for the genes indicated were queried from 279 head and neck squamous cell carcinomas from The Cancer Genome Atlas (TCGA) using cbioportal (http://www.cbioportal.org/public-portal/). TP53 is mutated in over 70 % of tumors. The adjacent loci containing TP63 and PI3K catalytic subunit PIK3CA are co-amplified in ~20 % of HNSCC, and activating mutations in PIK3CA are observed in additional tumors. Mutations in the oxidative stress pathway including KEAP1 mutations (~5 %) and amplification or mutation of NRF2 (~12 %) are found. Key, red bars, amplifications; blue bars, homozygous deletion; green bars, mutations
22.5 Role of KEAP1/NRF2 Genetic Alterations in HNSCC Susceptibility
KEAP1 is an important sensor of oxidative stress, and ubiquitin ligase, which in the absence of stress binds and promotes proteasomal degradation of IKKβ proteins, inhibiting NF-κB activation and cell survival, and of transcription factor NRF2, inhibiting antioxidant genes (Fig. 22.1) (Tian et al. 2012). In the presence of ROS, KEAP1 cysteine residues undergo conformational changes that promote IKK-induced NF-κB activation and cell survival, while releasing NRF2 for nuclear translocation and activation of antioxidant genes. The antioxidant genes include glutathione-S-transferases (GSTs), NADP(H) quinone oxidoreductase (NQO1), catalase, and superoxide dismutases (SODs), important in neutralizing ROS. In HNSCC, mutations of KEAP1 are observed in ~5 % and in NRF2 are observed in ~12 % of HNSCC (Fig. 22.2), suggesting genomic alterations affect KEAP1 regulated NF-κB prosurvival signaling and NRF2 antioxidant responses in a subset of HNSCC. Most HNSCC tumors with alterations in KEAP1 and NRF2 also appear to have undergone mutations in TP53 (Fig. 22.2). Studies in transgenic mouse models suggest NRF2 may inhibit initiation of tumorigenesis, but enhance progression of established tumors (Satoh et al. 2013). This observation involving NRF2 and endogenous antioxidants mirrors the cautionary observation that antioxidant β-carotene can inhibit initiation in preclinical models of lung cancer, while enhancing progression and mortality in smokers (ATBC 2003), who could have had premalignant lesions with TP53 mutations.
22.6 Role of Alterations in Fanconi/BRCA DNA Damage Response in HNSCC Susceptibility
The Fanconi Anemia (FANC) and Breast/ovarian cancer (BRCA) genes and proteins are now known to comprise a pathway critical in mediating repair of ROS-mediated DNA damage by nonhomologous recombination (Kee and D’Andrea 2012). Overall, the pathway includes 15 FANC genes, BRCA1 and BRCA2. Mutations in FANCA, FANCB, FANCC, FANCE, FANCF, FANCG, FANCL, and FANCM account for approximately 90 % of patients. These result in loss of FANCD2 and FANCI monoubiquitylation, the key regulatory event in the FA pathway. Besides loss of DNA repair, FANCD2 activates transcription of a TP53 homologue TAp63 that suppresses tumorigenesis (Park et al. 2013). Patients with FANC mutations are prone to bone marrow failure with anemia and leukemia in childhood, or development of HNSCC and genitourinary tract SCCs in young adulthood. While BRCA gene mutations predispose to breast and ovarian cancer, they have also been detected in patients with HNSCC. Overall, genomic alterations in FANC and BRCA genes are detected in 86/279 (~31 %) of HNSCC tumors in TCGA (Fig. 22.3). The HNSCC that arise in patients with FA patients in their 20–40 s frequently occur in the absence of tobacco use, and occur in the oral cavity in the tongue and mucosa adjacent to areas of exposure to dental trauma and microbiota (Van Waes 2005). The FA pathway has recently been shown to limit human papilloma virus replication and transformation by the HPV E7 gene (Hoskins et al. 2012; Park et al. 2010). However, the extent of the role of HPV in FA HNSCC remains unclear, as others report that the mutational spectrum in HNSCC in FA includes genes such as TP53, similar to that in tobacco-related HNSCC (van Zeeburg et al. 2008). Increased oxidative stress and potential for mutations and malignant transformation has been detected in FA cells (Du et al. 2008), and is enhanced by inflammatory signaling and induction of ROS by TNFα (Li et al. 2007).
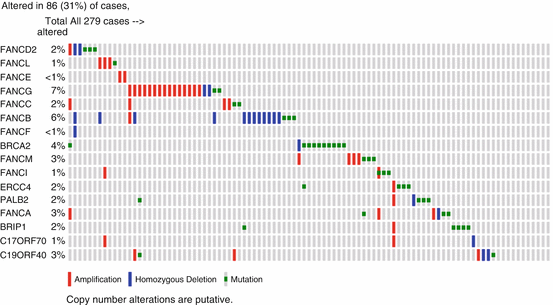
Fig. 22.3
Genomic alterations in Fanconi and BRCA genes in HNSCC. Publically available data for the genes indicated were queried from 279 head and neck squamous cell carcinomas from The Cancer Genome Atlas (TCGA) using cbioportal (http://www.cbioportal.org/public-portal/). Fanconi genes include named FANC, and other genes listed. Most genes exhibit deletions or inactivating mutations, but FANCG is more often amplified, which could enhance or repair of ROS-mediated genomic instability, respectively. Key, red bars, amplifications; blue bars, homozygous deletion; green bars, mutations
22.7 Potential of Anti-inflammatory Agents, Antioxidants, and PI3K-mTOR Inhibitors to Delay Malignant Progression and for Clinical Translation
Based on the potential role of HNSCC-associated inflammation and ROS in promoting HNSCC, anti-inflammatory drugs have been of interest. Many anti-inflammatory drugs inhibit NF-κB or NF-κB targets such as Cyclooxygenases, responsible for inflammatory prostaglandins (Van Waes 2007). Proteasome inhibitors preventing IκB degradation and NF-κB activation and inflammation yielded incomplete and transient responses in preclinical and clinical trials, which were found to be due to compensatory activation of other prosurvival signaling pathways (Allen et al. 2008; Chen et al. 2008). Cyclooxygenase inhibitor ketorolac inhibited inflammatory cells in response to HNSCC in preclinical studies, but showed a similar 30 % response rate as placebo in reducing leukoplakia (Hong et al. 2000; Mulshine et al. 2004). However, Cyclooxygenase 2 plus Epidermal Growth Factor Receptor inhibitors were found to synergistically inhibit head and neck squamous cell carcinoma tumorigenesis in preclinical and clinical studies (Saba et al. 2014). In a phase I study with a combination of COX2 inhibitor celecoxib and EGFR inhibitor erlotinib in patients with advanced premalignant lesions, the overall histologic response rate was 63 % (complete response 43 %, partial response 14 %, stable disease 29 %, disease progression 14 %). With median follow-up of 36 months, mean time to progression to higher-grade dysplasia or carcinoma was 25.4 months. Encouraging responses to the celecoxib and erlotinib combination correlated with EGFR pathway inhibition, where downregulation of EGFR and p-ERK in follow-up biopsies correlated with response to treatment (Vander Broek et al. 2013).
With evidence for a relatively high prevalence of PI3K-mTOR pathway alterations HNSCC, and their importance in activation of NF-κB and inflammatory responses (Vander Broek et al. 2013), PI3K and mTOR inhibitors have been the subject of preclinical and clinical investigation. In genetically engineered experimental animal models with genetic defects in TGFβ receptor 1/Pten genes and activated PI3K signaling, a synthetic PI3K-mTOR inhibitor delayed onset of HNSCC, demonstrating clinical translational potential (Herzog et al. 2013). In a clinical trial of mTOR inhibitor rapamycin underway at NIH, clinical responses have been observed in patients with stage II–IV oral and oropharyngeal cancers [C. Van Waes, unpublished observations].
Based on the hypothesis that FA cells are more prone to oxidative damage, we examined and demonstrated an increase in ROS DNA marker 8-OHdG in human FA fibroblast lines relative to control cell lines (Zhang et al. 2008). A synthetic nitrosamine antioxidant tempol reduced 8-OHdG similar to normal levels in these FA cells, and cells from Fancd2 knockout mice. Fancd2−/− Trp53+/− mice on a tempol diet showed a significantly longer mean tumor-free survival (mean = 390 days) than the mice on placebo diet (mean = 308 days) (P < 0.01). After early deaths due to leukemias, statistical analysis revealed that tempol treatment significantly increased the mean epithelial tumor-free survival time by 38 % in Fancd2−/− Trp53+/− mice (P < 0.0001). These data suggest that tempol may have a role in reducing oxidative DNA damage and malignant transformation in FA (Zhang et al. 2008), although naturally occurring antioxidant resveratrol or n-acetylcysteine did not have significant chemopreventive effects in the same model (Zhang et al. 2014).
In conclusion, anti-inflammatory, antioxidants, and PI3K-mTOR inhibitors targeting specific genetic alterations have preclinical or clinical activity and potential for further clinical investigation in prevention of HNSCC.
Acknowledgement
Supported by NIDCD intramural research projects ZIA-DC-000016, -73, and -74.
References
Allen C, Saigal K, Nottingham L, Arun P, Chen Z, Van Waes C (2008) Bortezomib-induced apoptosis with limited clinical response is accompanied by inhibition of canonical but not alternative nuclear factor-{kappa}B subunits in head and neck cancer. Clin Cancer Res 14:4175–4185. doi:10.1158/1078-0432.CCR-07-4470 PubMedCrossRef
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


