Chapter 20 Regulatory affairs
Brief history of pharmaceutical regulation
As described in Chapter 1, the development of experimental pharmacology and chemistry began during the second half of the 19th century, revealing that the effect of the main botanical drugs was due to chemical substances in the plants used. The next step, synthetic chemistry, made it possible to produce active chemical compounds. Other important scientific developments, e.g. biochemistry, bacteriology and serology during the early 20th century, accelerated the development of the pharmaceutical industry into what it is today (see Drews, 1999).
This catastrophe became a strong driver to develop animal test methods to assess drug safety before testing compounds in humans. Also it forced national authorities to strengthen the requirements for control procedures before marketing of pharmaceutical products (Cartwright and Matthews, 1991).
Another blow hit Japan between 1959 and 1971. The SMON (subacute myelo-optical neuropathy) disaster was blamed on the frequent Japanese use of the intestinal antiseptic clioquinol (Entero-Vioform®, Enteroform® or Vioform®). The product had been sold without restrictions since early 1900, and it was assumed that it would not be absorbed, but after repeated use neurological symptoms appeared, characterized by paraesthesia, numbness and weakness of the extremities, and even blindness. SMON affected about 10 000 Japanese, compared to some 100 cases in the rest of the world (Meade, 1975).
European (EEC) efforts to harmonize requirements for drug approval began 1965, and a common European approach grew with the expansion of the European Union (EU) to 15 countries, and then 27. The EU harmonization principles have also been adopted by Norway and Iceland. This successful European harmonization process gave impetus to discussions about harmonization on a broader international scale (Cartwright and Matthews, 1994).
International harmonization
ICH conferences, held every 2 years, have become a forum for open discussion and follow-up of the topics decided. The important achievements so far are the scientific guidelines agreed and implemented in the national/regional drug legislation, not only in the ICH territories but also in other countries around the world. So far some 50 guidelines have reached ICH approval and regional implementation, i.e. steps 4 and 5 (Figure 20.1). For a complete list of ICH guidelines and their status, see the ICH website (website reference 1).
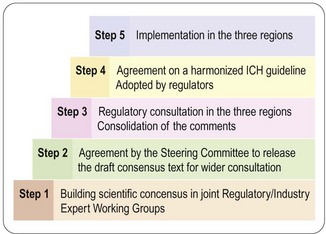
The process described in Figure 20.1 is very open, and the fact that health authorities and the pharmaceutical industry collaborate from the start increases the efficiency of work and ensures mutual understanding across regions and functions; this is a major factor in the success of ICH.
Roles and responsibilities of regulatory authority and company
 approves clinical trial applications
approves clinical trial applications
 gives procedural and scientific advice to companies during drug development
gives procedural and scientific advice to companies during drug development



 owns the documentation that forms the basis for assessment, is responsible for its accuracy and correctness, for keeping it up to date, and for ensuring that it complies with standards set by current scientific development and the regulatory authorities
owns the documentation that forms the basis for assessment, is responsible for its accuracy and correctness, for keeping it up to date, and for ensuring that it complies with standards set by current scientific development and the regulatory authorities

 has a right to appeal and to correct cases of non-compliance.
has a right to appeal and to correct cases of non-compliance.
The drug development process
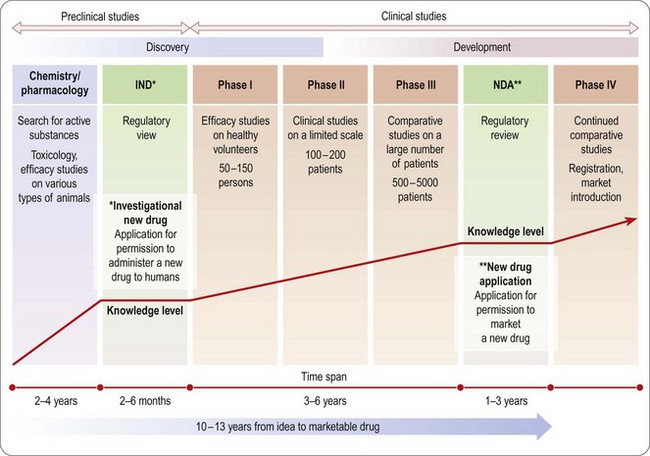
An overview of the process of drug development is given in Chapters 14–18 and summarized in Figure 20.2. As already emphasized, this sequential approach, designed to minimize risk by allowing each study to start only when earlier studies have been successfully completed, is giving way to a partly parallel approach in order to save development time.
Safety assessment (pharmacology and toxicology)
Next we consider how to design and integrate pharmacological and toxicological studies in order to produce adequate documentation for the first tests in humans. ICH guidelines define the information needed from animal studies in terms of doses and time of exposure, to allow clinical studies, first in healthy subjects and later in patients. The principles and methodology of animal studies are described in Chapters 11 and 15. The questions discussed here are when and why these animal studies are required for regulatory purposes.
Primary pharmacology
The primary pharmacodynamic studies provide the first evidence that the compound has the pharmacological effects required to give therapeutic benefit. It is a clear regulatory advantage to use established models and to be able at least to establish a theory for the mechanism of action. This will not always be possible and is not a firm requirement, but proof of efficacy and safety is helped by a plausible mechanistic explanation of the drug’s effects, and this knowledge will also become a very powerful tool for marketing. For example, understanding the mechanism of action of proton pump inhibitors, such as omeprazole (see Chapter 4), was important in explaining their long duration of action, allowing once-daily dosage of compounds despite their short plasma half-life.
General pharmacology
General pharmacology1 studies investigate effects other than the primary therapeutic effects. Safety pharmacology studies (see Chapter 15), which must conform to good laboratory practice (GLP) standards, are focused on identifying the effects on physiological functions that in a clinical setting are unwanted or harmful.
Pharmacokinetics: absorption, distribution, metabolism and excretion (ADME)
Preliminary pharmacokinetic tests to assess the absorption, plasma levels and half-life (i.e. exposure information) are performed in rodents in parallel with the preliminary pharmacology and toxicology studies (see Chapter 10).
Toxicology
The principles and methodology of toxicological assessment of new compounds are described in Chapter 15. Here we consider the regulatory aspects.
Single and repeated-dose studies
The acute toxicity of a new compound must be evaluated prior to the first human exposure.
This information is obtained from dose escalation studies or dose-ranging studies of short duration. Lethality is no longer an ethically accepted endpoint. The toxicology requirements for the first exploratory studies in man are described in the ICH guidline M3 which reached step 5 in June 2009 (see website reference 1). Table 20.1 shows the duration of repeated-dose studies recommended by ICH, to support clinical trials and therapeutic use for different periods.

Genotoxicity
Preliminary genotoxicity evaluation of mutations and chromosomal damage (see Chapter 15) is needed before the drug is given to humans. If results from those studies are ambiguous or positive, further testing is required. The entire standard battery of tests needs to be completed before Phase II (see Chapter 17).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


