particularly for a drug that has limited distribution and use (such as an orphan drug for a rare disease). That issue is often complex because a larger number of patients with heterogeneous characteristics take a new drug after it is marketed than during its investigational period. Therefore, postmarketing data in the form of a registry provide important information to better understand the drug’s benefit-to-risk relationship.
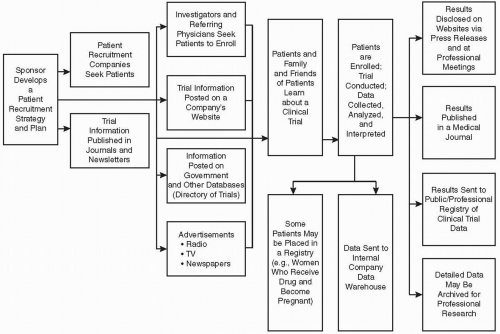 Figure 83.1 Schematic illustration of how a company’s recruitment strategy leads to dissemination of information about a trial and reaches patients, via directories and other methods. The results of a clinical trial are disseminated via a variety of methods and approaches, including a proposed registry available to the public. Inclusion of the data in regulatory submissions is not shown. |
will be entered in the registry, and their clinical outcome during hospitalization and at one and six months after discharge will be assessed. Many additional patient registries can be readily found through the National Institutes of Health PubMed database (www.pubmed.gov) as well as many others.
and is encouraged (but is not mandatory) for other trials. Listing the trial is free. CenterWatch charges fees for listing trials on their website (www.centerwatch.com). Individual companies also list on their own websites many of the trials they are sponsoring.
| ||||||||||||||||||||||||||||||||||||||||||
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


