Fig. 1
TRP channels expressed on the peripheral terminals of sensory neurons as molecular targets for itch- and pain-producing agents. Please note that TRPV1 is a direct downstream target for histamine. TRPV1 is also expressed on neurons that mediate non-histaminergic itch: some pruritogens (e.g., proteases) indirectly activate TRPV1 (via PAR-2), whereas others (e.g., chloroquine acting through TRPA1) bypass TRPV1. TRP channel expression is, however, not specific for itch-transmitting afferents: nociceptive neurons also express an overlapping subset of TRP channels. Observe that at the level of the spinal cord, nociceptive neurons exert a tonic inhibition (via Bhlhb5-expressing neurons) on the pruriceptive pathway. This model postulates that itch and pain are transmitted by distinct afferents with overlapping TRP channel expression. Abbreviations are ET-1 endothelin-1, 5-HT 5-hydroxytryptamine, GPCR G protein-coupled receptor, TLR toll-like receptor, Mrgpr Mas-related G protein-coupled receptor, PAR2 protease-activated receptor-2, H1/H4 histamine receptor 1 and 4, PLC phospholipase C, PIP2 phosphatidylinositol 4,5-bisphosphate, SP substance P, VGLUT2 vesicular glutamate transporter 2, NPPB natriuretic peptide type-B, GRP gastrin-releasing peptide, GABA gamma-aminobutyric acid, Bhlhb5 basic loop helix transcription factor, beta-3 subfamily-5
3.1 Histamine
Histamine is the central mediator of local (cutaneous) inflammatory reactions and systemic allergic responses. It is mostly released from mast cells and basophils following activating stimuli (Metz and Maurer 2009; Metz et al. 2008). Arguably, histamine is the best known itch-producing substance (Jeffry et al. 2011; Metz et al. 2011). Although histamine can evoke itch by directly activating sensory neurons, its role in the pathogenesis of pruritic skin diseases is yet to be convincingly demonstrated. Indeed, although antihistamines provide relief in urticarial diseases (Ortonne 2012; Zuberbier and Maurer 2007), most pruritic chronic skin disorders (e.g., AD) are resistant to antihistamine therapies (Jeffry et al. 2011; Klein and Clark 1999; Metz et al. 2011). In accord, in a mouse model of AD, histamine failed to evoke scratching behavior (Akiyama et al. 2010).
Histamine receptors belong to the G protein-coupled receptor (GPCR) superfamily. Of the four known histamine receptor subtypes (H1–H4) (Jeffry et al. 2011), two (H1 and H4) are believed to function as pruriceptors (Bell et al. 2004; Jeffry et al. 2011). Unexpectedly, H3 receptor inverse agonist and antagonists were reported to induce scratching behavior in mice (Hossen et al. 2006; Rossbach et al. 2011), whereas H3 agonists were shown to attenuate the symptom of allergic rhinitis (Yokota et al. 2008). These findings might be explained by the expression of the inhibitory H3 receptor on the sensory nerve endings. These receptors could inhibit substance P (SP) release from sensory terminals (Arrang et al. 1983; Ohkubo et al. 1995; Rossbach et al. 2011; Yokota et al. 2008) and disrupt the sensory nerve–mast cell “itchy circuit” (see below). If this hypothesis holds true, the intimate relationship of histamine to itch might be more complicated than previously thought.
3.2 Proteases and Their Receptors
Serine proteases (including trypsin, chymotrypsin, chymase, and kallikrein) are capable of activating proteinase-activated receptors (PARs) (Steinhoff et al. 2005; Vu et al. 1991). Of PARs, PAR-2 (expressed on sensory neurons) is the major itch mediator (Steinhoff et al. 2000, 2003a; Vergnolle et al. 2003). For example, cowhage spicules contain the protease mucunain that can activate PAR-2. When applied to the human skin, cowhage evokes a histamine-independent itch reaction (Davidson and Giesler 2010; Davidson et al. 2007; Papoiu et al. 2011, 2012; Reddy et al. 2008). In AD patients, a dysregulation of PAR-2 was observed both in sensory fibers and in epidermal keratinocytes (Buddenkotte et al. 2005). In addition, kallikrein activity was increased in pruritic papular eruptions (Reis et al. 1999). In Netherton syndrome, mutations in the serine protease inhibitor Kazal-type 5 can lead to increased PAR-2 stimulation due to epidermal protease hyperactivity; this mechanism may be responsible for the development of AD-like symptoms in these patients (Briot et al. 2009).
3.3 Neuropeptides and Neurotrophins
Sensory neuropeptides released from peripheral terminals of activated sensory neurons can initiate the release of pruritogens from non-neuronal cell types of the skin. For example, the neurokinin SP, a well-known pain mediator, is also able to induce the degranulation of mast cells (Church et al. 1991; Kulka et al. 2008; Thomsen et al. 2002; van der Kleij et al. 2003). [Parenthetically, the mast cell SP receptor is different from the neurokinin NK1 receptor.] In addition to histamine, mast cells also release various proinflammatory mediators which, in turn, may further increase the activity of sensory neurons, creating a positive feedback (an “itchy circuit”) (Ansel et al. 1997; Luger 2002; Paus et al. 2006a, b; Steinhoff et al. 2003b). As the initiator of this feedback loop, SP can be considered as an important etiological factor in various “itchy” skin diseases. Indeed, AD patients show significantly elevated plasma SP levels (Salomon and Baran 2008). Furthermore, in prurigo nodularis lesions increased density of SP+ nerve fibers was found compared to non-affected skin areas (Abadia Molina et al. 1992; Haas et al. 2010). Patients with hepatic (cholestasis-associated) pruritus also have elevated plasma SP levels (Trivedi and Bergasa 2010).
The pruriceptive role of CGRP remains controversial. The findings are conflicting. Some studies suggested that CGRP prolonged the latency of SP-induced itch (Ekblom et al. 1993). In keeping with this, decreased CGRP plasma levels were found in AD patients during the exacerbation period. By contrast, significantly higher CGRP values were measured in patients with severe pruritus compared to those with mild pruritus (Salomon and Baran 2008). Increased CGRP levels were also reported in patients with pruritic dermatoses (e.g., AD, nummular eczema, prurigo nodularis) (Jarvikallio et al. 2003; Liang et al. 2000). In anesthetized mice, scratching itself increased the outgrowth of CGRP+ nerve fibers (Yamaoka et al. 2007). Based on these results, one may speculate that decreased CGRP level might be a primary itch-inducing factor, whereas the elevation of CGRP might only be a consequence of scratching.
Neurotrophins (such as nerve growth factor [NGF], neurotrophin-3 and neurotrophin-4, and glial cell line-derived neurotrophic factor [GDNF]) are well-known regulators of cutaneous nerve development and regeneration (Botchkarev et al. 2006). Neurotrophins may also play an etiological role in the development of itch sensation and pruritic diseases. Several cell types of the skin (e.g., keratinocytes, mast cells, and fibroblasts) produce and release neurothrophins (Groneberg et al. 2005). Neurotrophins significantly influence itch sensation in various pathophysiological conditions via modulating the innervation density of the skin, as well as regulating the expression and/or activity of receptors expressed by sensory neurons. For example, expression of NGF is highly increased by inflammation and injuries; this, in turn, initiates acute sensitization and sprouting (leading to chronic sensitization) of C-type afferent fibers via activating specific TrkA receptors (Botchkarev et al. 2006; Hefti et al. 2006; Lazar et al. 2004; Urashima and Mihara 1998). Increased expression and plasma level of NGF were also reported in pruritic skin diseases such as in AD (Toyoda et al. 2002), prurigo nodularis (Johansson et al. 2002), and psoriasis vulgaris (Nakamura et al. 2003). Moreover, increased neurotrophin-4 expression was found in lesional skin of AD patients (Grewe et al. 2000).
Importantly, NGF levels correlated to the symptom severity in AD, and they declined with effective treatment (Yamaguchi et al. 2009). In accord with these findings, atopic human keratinocytes cocultured with porcine sensory neurons were shown to induce increased neurite outgrowth. This effect was mediated by NGF and GDNF (Roggenkamp et al. 2012). NGF not only stimulated the sprouting of itch-sensitive C-fibers but also upregulated the expression of SP, CGRP (Chang et al. 2007; Raychaudhuri and Raychaudhuri 2004; Roggenkamp et al. 2012; Verge et al. 1995), and TRPV1 (Amaya et al. 2004; Ji et al. 2002; Zhang et al. 2005b). Combined, these changes promote itch sensation. Last, neurotrophins may also directly induce degranulation of mast cells with resultant histamine release (Marshall et al. 1999; Metz et al. 2004; Stempelj and Ferjan 2005; Tal and Liberman 1997).
3.4 Inflammatory Mediators as Peripheral Itch Sensitizers
Nociceptive sensory neurons can be sensitized by an array of inflammatory mediators produced during acute or chronic inflammation of the skin, a phenomenon called inflammatory hyperalgesia (Reeh and Kress 1995). Various cell types of the skin are involved in the generation of these mediators. For example, (1) mast cells release histamine, tryptase, serotonin, tumor necrosis factor-α (TNF-α), leukotrienes, and prostaglandins; (2) keratinocytes synthesize prostaglandins, interleukins, and neurotrophins; (3) sebocytes produce prostaglandins, leukotrienes, and interleukins; (4) endothelial cells generate kinins and endothelins; and (5) immune cells contribute interleukins and chemokines. Importantly, the majority of these mediators possess marked pruritogenic potentials (Bickford 1938; Biro et al. 2005; Ikoma et al. 2006; Paus et al. 2006a; Stander et al. 2003; Steinhoff et al. 2006; Toth et al. 2011).
The prostaglandins PGE1 and PGE2 were shown to initiate and/or potentiate itch responses independent of histamine liberation (Boss and Burton 1981; Hägermark and Strandberg 1977; Lovell et al. 1976; Neisius et al. 2002). In addition, intradermal injection of other eicosanoids, such as leukotriene B4 (LTB4) or a stable thromboxane A2 analogue, evoked itch-associated responses in mice (Andoh and Kuraishi 1998; Andoh et al. 2007). Furthermore, PAR-2 activation in keratinocytes resulted in LTB4 (and PGE2) release in vitro and evoked scratching behavior in vivo. These effects were reduced by 5-lipoxygenase inhibition, suggesting that LTB4 may mediate the effect of PAR-2 activation (Zhu et al. 2009). Interestingly, LTB4 is also thought to play a role in mediating SP-evoked itch (Andoh et al. 2001).
By contrast, PGD2 exerts an antipruritic effect (Kaur et al. 2010; Sugimoto et al. 2007). In accord, PGD2 can suppress IgE-induced histamine release from the RBL-2H3 mast cell line (Hashimoto et al. 2005). Both pharmacological inhibition and in vivo silencing of cyclooxygenase-1 (COX1) resulted in decreased PGD2 levels and enhanced the scratching behavior in the AD model, NC/Nga mice (Inoue et al. 2007; Sugimoto et al. 2006).
Bradykinin also induces itch (Cormia and Dougherty 1960; Hägermark 1974; Schmelz et al. 2003) by (1) enhancing histamine release from mast cells (Walker et al. 1995), (2) augmenting histamine-evoked responses (Koppert et al. 2001; Lang et al. 1990), and (3) sensitizing the sensory afferent fibers and their receptors (e.g., TRPV1) (Lang et al. 1990). Bradykinin applied to the skin enhances the release of neuropeptides and prostaglandins (Averbeck and Reeh 2001) and evokes itch on lesional atopic skin via a histamine-independent manner (Hosogi et al. 2006). Both B1 and B2 bradykinin receptors were shown to mediate protease and PAR-2 activation-evoked scratching in mice (Costa et al. 2008, 2010). Bradykinin was also found to evoke scratching responses in complete Freund’s adjuvant (FCA)-inflamed skin (Liang et al. 2012).
Immune cells of the skin can produce a variety of cytokines and interleukins (ILs) which may contribute to the pathogenesis pruritic disorders. For example, IL-2 was found to induce itch (Gaspari et al. 1987; Wahlgren et al. 1995) by activating a histamine- and bradykinin-sensitive subpopulation of sensory C-fibers (Darsow et al. 1997; Martin and Murphy 1995). Interestingly, bradykinin potentiated the responsiveness of polymodal nociceptors to IL-2 (Martin 1996), resulting in a bidirectional augmentation. IL-2 appears to be clinically relevant in that elevated IL-2 serum levels were reported in hemodialyzed patients with uremic pruritus compared to non-pruritic individuals (Fallahzadeh et al. 2011; Martin 1996). Of note, in mice cutaneous overexpression of IL-4 resulted in an AD-like pruritic inflammatory skin disease (Chan et al. 2001). Furthermore, increased IL-6-like immunoreactivity was reported in cutaneous nerve fibers of prurigo nodularis patients (Nordlind et al. 1996).
Recently, additional cytokines (e.g., IL-13 and IL-31) have been linked to AD. Transgenic overexpression of IL-31, as well as skin-specific overexpression of IL-13, resulted in severe pruritic dermatitis in mice (Dillon et al. 2004; Zheng et al. 2009). Furthermore, both AD patients (Bilsborough et al. 2006; Nobbe et al. 2012) and atopic mice (Takaoka et al. 2005) showed increased expression of IL-31, with levels correlating to symptom severity and scratching behavior in humans and mice, respectively (Ezzat et al. 2011; Kim et al. 2011a; Sonkoly et al. 2006). Although IL-31 seems to be an important player in AD (and intradermal IL-31 injection evokes itch both in NC/Nga and BALB/c mice (Arai et al. 2013), the exact mechanism by which IL-31 contributes to itch sensation remains unclear. Of note, in the skin, IL-31 is mostly released by Th2 lymphocytes and mature dendritic cells (Cevikbas et al. 2013), and IL-31 receptors are also expressed both on a subset of DRG neurons (Bando et al. 2006; Cevikbas et al. 2013) and on normal human epidermal keratinocytes (NHEKs) (Kasraie et al. 2011). IL-31 receptor expression on NHEKs is increased by stimulation of the toll-like receptor, TLR-2. This, in turn, results in enhanced secretion of the proinflammatory chemokine ligand, CCL2 (also known as monocyte chemotactic protein-1). Intriguingly, this effect is absent in epidermal keratinocytes obtained from AD patients where TLR-2 expression is impaired (Kasraie et al. 2011). Finally, it should be mentioned here that in pruritic myeloproliferative disorders mast cells release (among others) increased amounts of IL-31 (Ishii et al. 2009).
4 The “Pruritic” TRP Channels
TRP channels were initially discovered in a blind strain of Drosophila (Cosens and Manning 1969): when exposed to prolonged intense light, these mutant fruit flies showed transient calcium influx into their photoreceptor cells (Montell and Rubin 1989), hence the term “transient receptor potential”(Minke 2010). This seminal observation paved the way to the discovery of the first mammalian TRP channels called “canonical” (TRPC) due to their homology to the Drosophila channels (Wes et al. 1995; Zhu et al. 1995). Unlike most families of ion channels that have been grouped together based on function, TRP channels are subdivided based on primary amino acid structure (Clapham et al. 2001; Nilius and Voets 2005; Pedersen et al. 2005; Wu et al. 2010). As of today, mammalian TRP channels comprise 28 members and are categorized into six subfamilies: TRPC (canonical), TRPV (vanilloid), TRPM (melastatin), TRPP (polycystin), TRPML (mucolipin), and TRPA (ankyrin). The mucolipin and polycystin subfamilies were named after the diseases (“TRP channelopathies”) they are associated with, mucolipidosis and autosomal dominant polycystic kidney disease, respectively (Nilius et al. 2007). The vanilloid subfamily was named after its founding member, the vanilloid (capsaicin) receptor TRPV1 (Caterina et al. 1997). The first melastatin channel (TRPM1) was discovered as a protein present in benign nevi and absent in malignant melanoma (Duncan et al. 1998). As of today, the ankyrin subfamily has only one member, TRPA1, which, as the name implies, is rich in ankyrin segments at its N-terminus.
As a general rule, TRP channels have six transmembrane-spanning domains (S1 to S6) with a pore-forming loop between S5 and S6 (Wu et al. 2010). Most TRPs form functional channels as homotetramers, but heteromultimerization is frequently observed (Cheng et al. 2010a). Beyond that, few generalities can be made about TRP channels. Consistent with the diversity in primary structure is the diversity observed in functional properties. The TRPs are commonly referred to as calcium-permeable nonselective cation channels, but TRPM4 and TRPM5 are sodium-selective channels, whereas TRPV5 and TRPV6 are inwardly rectifying calcium-selective channels (Owsianik et al. 2006). TRP channels also vary in their tissue expression patterns. For example, TRPM7 is ubiquitously expressed, whereas the expression of TRPA1 is highly restricted. Even the subcellular localization of the proteins varies with family members: for example, TRPML1 is being hypothesized to be an intracellular channel (Bach 2005). Some TRP channels like TRPM2 are unique in that they contain a functional NUDIX/ADP ribose domain along with a kinase domain that bears some resemblance to protein kinase A (Eisfeld and Luckhoff 2007). These TRPs combine features of ion channels and enzymes and are therefore referred to by some as “chanzymes” (Montell 2003).
Despite decades of intensive research, only a few endogenous ligands of TRP channels have been identified. For example, some endogenous lipid mediators, like arachidonic acid derivatives or lipoxygenase products, can activate some TRP channels like TRPV1 (Huang et al. 2002; Hwang et al. 2000; Vriens et al. 2009; Watanabe et al. 2003), whereas other TRPs (e.g., TRPM2) are believed to function as redox sensors (Kozai et al. 2013; Miller and Zhang 2011). In the days of the finalization of this manuscript, the first high-resolution TRP channel structure was published: this pioneering finding can potentiate the discovery of further endogenous ligands and their structure–activity relationships (Cao et al. 2013; Liao et al. 2013). For the time being, most TRP channels, however, remain orphan receptors. How these channels are modulated in vivo is still largely a mystery.
A functional subset of TRP channels (the so-called thermoTRPs) is distinguished by their sensitivity to temperature changes: combined, these channels cover a broad range of temperatures, ranging from noxious hot to ice-cold (Szallasi et al. 2007; Patapoutian et al. 2009; Talavera et al. 2008; Voets 2012; Voets et al. 2004). With respect to pruritus, these thermoTRPs (in particular, TRPV1, TRPV3, and TRPA1) are the most interesting (Figs. 1 and 2). However, some other members of the TRP family may also contribute to the development of itch and related diseases (Biro et al. 2007; Toth and Biro 2013).
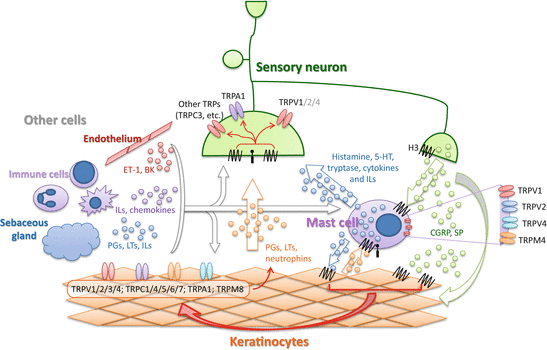
Fig. 2
A cartoon depicting the positive feedback loop between TRP channel-expressing neurons and non-neuronal cells, in particular keratinocytes and mast cells. Please note that both neuropeptides released from sensory afferents (e.g., SP and CGRP) and substances (PGs, LTs, and neurotrophins) generated by skin keratinocytes activate mast cells; in turn, mast cells release substances (histamine, 5-HT, tryptase, cytokines and ILs) that activate sensory afferents and stimulate keratinocytes. TRP channels that are expressed on sensory afferents, mast cells, and keratinocytes are believed to play a central role in these neuro-immune interactions. Abbreviations: PGs prostaglandins, ET-1 endothelin-1, BK bradykinin, ILs interleukins, LTs leukotrienes, SP substance P, CGRP calcitonin gene-related peptide
4.1 The Vanilloid (Capsaicin) Receptor TRPV1
Accumulating evidence suggests a crucial role for TRPV1 in pain sensation as a molecular integrator of a plethora of nociceptive stimuli. TRPV1 is a unique channel in that it functions as a polymodal nociceptor with a dynamic threshold of activation that could be significantly lowered under inflammatory conditions (reviewed in Szallasi et al 2007). Indeed, agents in an “inflammatory soup” act in concert to reduce the activation threshold of TRPV1. In addition to capsaicin, TRPV1 can also be activated and/or sensitized by numerous endogenous substances, including changes in pH [both acidosis (Caterina et al. 1997) and alkalosis (Dhaka et al. 2009)]; bradykinin (Chuang et al. 2001; Shin et al. 2002); ATP (Tominaga et al. 2001); lipoxygenase products (Hwang et al. 2000; Shin et al. 2002); prostaglandins (Mohapatra and Nau 2003); various neurotrophins such as NGF, neurotrophin-3, and neurotrophin-4 (Lazar et al. 2004; Shin et al. 2002); tumor necrosis factor-α (TNFα) (Nicol et al. 1997); and proinflammatory chemokines (Zhang et al. 2005a). In addition, activation of PAR-2 (Amadesi et al. 2004; Dai et al. 2004), metabotropic receptor-coupled hydrolysis of phosphatidylinositol 4,5-bisphosphate (PIP2) by phospholipase C (PLC) (Prescott and Julius 2003), or activation of the ionotropic purinoceptor P2X3 (Saloman et al. 2013) can indirectly lead to TRPV1 activation/sensitization. Some of these agents (like pH) activate TRPV1 directly, whereas others (like bradykinin) influence the phosphorylation state of the protein via protein kinases. There is preliminary evidence that TRPV1 can be operated by voltage, and the size of the pore is dynamically changing during activation (Chung et al. 2008; Voets 2012; Voets et al. 2004). Importantly, a subset of TRPV1-expressing neurons is activated by histamine (Kim et al. 2004; Shim et al. 2007). It has been shown that increases in cutaneous temperatures or exceedingly acidic pH of the skin (known TRPV1 activators) can effectively modulate itch sensation in humans (Paus et al. 2006a; Pfab et al. 2006; Steinhoff et al. 2006; Yosipovitch et al. 2005).
Several lines of evidence suggest that (similar to its role in nociception) TRPV1 may function as a molecular integrator of pruriceptive molecules in the itch pathway (Biro et al. 2005, 2007; Paus et al. 2006a; Steinhoff et al. 2006). Indeed, histamine-induced inward currents in sensory neurons can be blocked by TRPV1 antagonists (Shim et al. 2007). Activation by histamine of TRPV1-expressing neurons is mediated by the synthesis of 12-hydroperoxyeicosatetraenoic acid (12-HPETE), a pruritogenic (Kim et al. 2007) metabolite of 12-lipoxygenase and an endogenous activator of TRPV1 (Hwang et al. 2000; Shim et al. 2007). Moreover, upstream of TRPV1, the phospholipase A2 (PLA2)-coupled signaling pathway, was shown to mediate histamine-induced experimental itch (Kim et al. 2004; Shim et al. 2007). Recently, Imamachi and colleagues (2009) provided evidence for the role of phospholipase C beta3 (PLCβ3) in the downstream signaling pathway that links the histamine receptor to TRPV1. Indeed, histamine-induced scratching behavior was significantly reduced in TRPV1-deficient mice (Imamachi et al. 2009; Shim et al. 2007).
TRPV1 is clearly required for the transmission of histamine-induced scratching behavior but not for serotonin- or endothelin-1 (ET-1)-induced itch. However, chemical ablation by capsaicin of TRPV1-positive sensory neurons leads to deficits in behavioral responses to a variety of itch inductors, suggesting that the molecular targets for these molecules (though distinct from TRPV1) are also present on capsaicin-sensitive neurons (Imamachi et al. 2009).
The pharmacological blockade or genetic deletion of TRPV1 inhibited trypsin-evoked itch in mice (Costa et al. 2008), implying a role for TRPV1 in PAR-2-coupled pruriceptive signaling. In a rat model of hepatic pruritus, not only the expression of TRPV1 was increased in sensory neurons, but the expression of the channel shifted from the small- toward the medium-sized DRG neurons (Belghiti et al. 2013). The pruritogenic substance IL-31 also activated a small subset of TRPV1-positive neurons co-expressing the IL-31 receptor IL-31RA (Cevikbas et al. 2013). Approximately two thirds of the IL-31-responsive DRG neurons responded to capsaicin, and the majority (~90 %) were activated by mustard oil, an activator of both TRPV1 and TRPA1 (Everaerts et al. 2011). IL-31-evoked scratching behavior was significantly reduced both in TRPV1 and TRPA1 KO animals, suggesting that both molecules play a role in the IL-31-induced itch (Cevikbas et al. 2013). Unexpectedly, chloroquine (a molecule which, as discussed later, causes scratching behavior in mice via TRPA1 signaling) was also found to sensitize TRPV1 utilizing the MrgprA3–phospholipase C–PKC pathway (Than et al. 2013). Moreover, selective silencing of TRPV1+ sensory neurons ameliorates both histamine- and chloroquine-evoked responses (Roberson et al. 2013), providing further proof that TRPV1 can be involved in the mediation of both histaminergic and non-histaminergic itch.
The membrane-associated phosphoinositide-binding protein, Pirt, was recently identified as an important positive regulator of TRPV1 (Kim et al. 2008). Pirt-null mice showed not only impaired responsiveness to noxious heat and capsaicin (Kim et al. 2008) but also a decreased scratching response to both histaminergic and non-histaminergic (e.g., evoked by chloroquine, alpha-methyl-serotonin, ET-1, or PAR-2 activation) itch (Patel et al. 2011). Although the authors speculated that the itch-mediating effect of Pirt may be partly independent of TRPV1, it did not affect formalin-induced TRPA1 activation and the resultant scratching response (Patel et al. 2011). Of note, Pirt is not selective for TRPV1: indeed, it can regulate other TRP channels like TRPM8 (Tang et al. 2013).
Toll-like receptors (TLRs), originally identified as pathogen-pattern recognition receptors, are emerging as intriguing new players in itch sensation (Liu et al. 2012b). TLR7 is co-localized with TRPV1 in mouse sensory neurons where it mediates the imiquimod (a TLR7 agonist)-evoked scratch response. Interestingly, in TLR7-null mice the non-histaminergic itch response evoked by serotonin and ET-1 and PAR-2 activation or chloroquine was impaired. The chemical ablation of TRPV1-expressing DRG neurons by resiniferatoxin (an ultrapotent capsaicin analogue) treatment abolished the imiquimod response, but the inactivation of TRPV1 by genetic manipulation had no effect. Taken together, these findings imply a crucial role for TRPV1+ sensory afferents, but not the TRPV1 channel itself, in mediating TLR7-induced itch (Liu et al. 2010a).
The findings on the role of TLR7 in pruritus are, however, conflicting. Another study (Kim et al. 2011b) concluded that the pruritic effect of imiquimod was not related to TLR7, yet the authors also confirmed the role of TRPV1-expressing neurons (but, again, not of TRPV1) in the itch response. TLR3 (present in a subset of sensory neurons that co-express TRPV1 with GRP) was also reported to mediate both histamine-dependent and histamine-independent itch. Importantly, genetic deletion of TLR3 attenuated itch responses, but it did not affect acute pain sensation. In these animals, the responsiveness of DRG neuronal cell bodies was normal to both pain- and itch-producing agents, but the excitatory synaptic transmission in the spinal cord was impaired (Liu et al. 2012a).
Conditioned genetic deletion of the vesicular glutamate transporter type 2 (VGLUT2) in DRG neurons resulted in a dramatic increase in itch-related behavior, and, at the same time, it caused a significant deficiency in thermal pain sensation. The absence of VGLUT2 affected both the majority of TRPV1-expressing neurons that are negative for GRP (thought to be responsible for the pain transmission) and a smaller, partially overlapping subpopulation of TRPV1+ neurons that co-express GRP (which are suggested to be responsible for itch sensation). It was proposed that VGLUT2 deletion terminated the negative control that the pain-processing pathway exerts over the itch-processing pathway; this, in turn, resulted in a dramatic increase in spontaneous as well as histamine-dependent and histamine-independent scratching behavior. Indeed, in the VGLUT2-deficient mice, capsaicin evoked mostly itch at doses at which it caused pain in the wild-type littermate controls (Lagerstrom et al. 2010; Liu et al. 2010b). Most recently, in an elegant study, Han et al. (Han et al. 2013) reexpressed TRPV1 only in the MrgprA3-expressing neurons of TRPV1-null animals: in these animals, capsaicin evoked only itch but not pain responses. Combined, these findings provide strong experimental support for the selectivity theory.
The ongoing controversy that surrounds the function (or even very existence) of TRPV1 in non-neuronal cells is beyond the scope of this review. Here it suffices to mention that, albeit at a much lower level than in DRG neurons, widespread TRPV1 expression was reported in the human skin, including keratinocytes, dermal mast cells, dendritic cells, hair follicle (HF) keratinocytes, and sebocytes of the sebaceous glands (SG) (Basu and Srivastava 2005; Bodo et al. 2004, 2005; Denda et al. 2001; Inoue et al. 2002; Southall et al. 2003; Stander et al. 2004; Toth et al. 2009a, b). Moreover, TRPV1 activation (besides affecting the proliferation, differentiation, and apoptosis of keratinocytes) resulted in the release of cytokines and mediators (e.g., ILs, PGs, and growth factors) that may elicit itch sensation (Amantini et al. 2004; Basu and Srivastava 2005; Birder et al. 2001; Biro et al. 1998; Bodo et al. 2005; Southall et al. 2003; Toth et al. 2009b). TRPV1 expression is also increased in epidermal keratinocytes obtained from patients with prurigo nodularis (Stander et al. 2004). The expression of TRPV1 in skin mast cells is of particular interest (Bodo et al. 2004; Stander et al. 2004). One might speculate that TRPV1 (which is expressed on both sensory afferents and non-neuronal cells) may play a crucial bidirectional role in the interaction between neurons and non-neuronal cells that controls itch initiation and augmentation (Biro et al. 2005, 2007; Paus et al. 2006a; Steinhoff et al. 2006). Indeed, TRPV1 was reported to contribute to the LTB4-induced itch via mediating leukocyte infiltration as measured by myeloperoxidase (MPO) activity in rat dorsal skin (Fernandes et al. 2013).
If TRPV1-expressing sensory neurons play a central role in the development of pruriceptive itch, silencing of these neurons by pharmacological manipulation (e.g., capsaicin desensitization) should alleviate pruritus. Although there are anecdotal reports and case studies to report a beneficial effect for topical capsaicin application in different pruritic syndromes [ranging from notalgia paresthetica through psoriasis, prurigo nodularis, aquagenic pruritus, uremic pruritus, and cholestasis to pruritus ani and allergic rhinitis (Stander et al. 2001)] (Blom et al. 1997; Ellis et al. 1993; Lacroix et al. 1991; Lotti et al. 1994; Lysy et al. 2003; Makhlough et al. 2010; Stjarne et al. 1998; Tarng et al. 1996; Wallengren and Klinker 1995; Weisshaar et al. 2003), the antipruritic effect of topical capsaicin in controlled clinical trials is yet to be confirmed (Gooding et al. 2010). In dogs with a disease resembling human AD, itch-relieving effect was noted by the owners but not the investigators. A word of caution here: capsaicin-containing creams (e.g., Zostrix, 0.075 %) have been in clinical use for decades for the treatment of chronic painful conditions such as diabetic neuropathy. Despite their popularity, controlled clinical trials found no evidence that these creams have superior analgesic compared to placebo (Knotkova et al. 2008). To increase the exposure of cutaneous nerve endings to capsaicin, occlusive patches (Qutenza, 8 %) were developed (Noto et al. 2009). However, in 2012 the US Food and Drug Administration found no evidence that Qutenza relieves HIV-associated neuropathic pain. In summary, the clinical experience with topical capsaicin as an analgesic agent has been disappointing, and we have no reason to believe that topical capsaicin could fair much better in patients with chronic itch. The explanation why capsaicin desensitization has failed to live up to expectation is detailed elsewhere (Szallasi and Sheta 2012). A potential strategy to boost the efficacy of capsaicin desensitization is by using pore-permeable capsaicin analogs (e.g., permanently charged capsaicinoids) which target and desensitize only (hyper)active channels (Li et al. 2011; Moran et al. 2011).
After the cloning of TRPV1, there was a great deal of enthusiasm in the pharmaceutical industry to develop small molecule TRPV1 antagonists as analgesic agents (Szallasi et al. 2007). Indeed, a number of compounds entered clinical trials, but none has been advanced so far beyond Stage II due to a combination of unforeseen side effects (e.g., hyperthermia and impaired heat pain sensation) and lack of clinical efficacy. For further details, interested readers are referred to recent reviews (Brederson et al. 2013; Szallasi and Sheta 2012; Szolcsanyi and Pinter 2013; Trevisani and Szallasi 2010; Wong and Gavva 2009). Pilot studies with the TRPV1 antagonist PAC-14028 reported beneficial effects to alleviate itch in different AD models (Lim and Park 2012; Yun et al. 2011a, b). For example, in a mouse model of AD (induced by Dermatophagoides farina and oxazolone), PAC-14028 administration prevented the development of dermatitis-associated barrier damages by suppressing transepidermal water loss, inducing reconstruction of epidermal lipid layers, and normalizing altered expressions of epidermal differentiation markers and, at the same time, improved the AD-like symptoms (clinical severity, skin score, serum IgE levels, mast cell degranulation status). These beneficial effects obtained with TRPV1 antagonists need to be confirmed in AD patients, especially because SB-705498, another TRPV1 antagonist, did not provide any symptomatic relief in patients with seasonal allergic rhinitis (Bareille et al. 2013), although it was effective in guinea pig models (Changani et al. 2013; Delescluse et al. 2012).
The endocannabinoid anandamide (depending on its concentration and other local factors such as acidosis in inflamed tissue compartments (Olah et al. 2001) may also function as an endogenous TRPV1 agonist, that is, an “endovanilloid” (Biro and Kovacs 2009; Di Marzo et al. 2002; Di Marzo and Petrocellis 2006; Pacher et al. 2006). Indeed, it was suggested that TRPV1 doubles as an “ionotropic cannabinoid receptor.” Interestingly, various synthetic cannabinoids (e.g., HU210, WIN 55, 212–2) and certain endocannabinoids (e.g., N-palmitoyl ethanolamine) were shown to ameliorate both histamine-induced and disease-related pruritus (Dvorak et al. 2003; Gingold and Bergasa 2003; Stander et al. 2006a). This effect was accompanied by decreased neuropeptide release from the sensory endings (Dvorak et al. 2003). Furthermore, cannabinoid CB2 receptors have recently been identified on epidermal keratinocytes where they co-localize with TRPV1 (reviewed in (Biro et al. 2009)). The activation of keratinocyte CB2 resulted in the release of β-endorphin (Ibrahim et al. 2005), an analgesic and pruritic pro-opiomelanocortin substance. Taken together, these findings strongly support a role for the endocannabinoid system in the modulation of pruritoception, presumably via both TRPV1 and CB2. If this concept is corroborated by further experimental evidence, compounds that target both TRPV1 and CB2 may represent a novel strategy to relieve itch.
For the sake of completeness, it should be mentioned here that TRPV1 has been implicated in the puritic side effects of various drugs. For example, the antifungal agent clotrimazole was reported to activate both TRPV1 and TRPA1 (and inhibit TRPM8), and it was speculated that this might contribute to the burning–itching sensation that clotrimazole evokes in some patients (Meseguer et al. 2008). Moreover, retinoids (widely used to treat various skin diseases) can also activate TRPV1 and evoke nocifensive behavior (Yin et al. 2013). The antitumor agent paclitaxel was reported to increase TRPV1 expression in the skin (Hara et al. 2013). Burning itch is a well-documented side effect of topically applied calcineurin inhibitors (e.g., pimecrolimus and tacrolimus); this itch is accompanied by the release of SP and CGRP from primary sensory afferents (Stander et al. 2006b). Tacrolimus may influence the phosphorylation state of TRPV1 (Pereira et al. 2010).
4.2 TRPV2
TRPV2 was originally cloned from rodent brain as a “VR1-like protein” (VRL-1), and it was also described in a subset of medium- to large-diameter sensory neurons as a detector of noxious hot temperature (>52 °C) (Caterina et al. 1999). It is also expressed in C-fibers innervating the human skin (Axelsson et al. 2009) in co-localization with its closest relative, TRPV1 (Rutter et al. 2005). Furthermore, TRPV2 is expressed on human epidermal keratinocytes and the human mast cell line HMC1 (Radtke et al. 2011; Zhang et al. 2012a).
The role of the TRPV2 in the noxious heat sensation is controversial. Indeed, TRPV2 KO mice display normal thermal and mechanical nociception (Park et al. 2011). Moreover, unlike its murine homologue, the human TRPV2 is insensitive to noxious heat (Neeper et al. 2007). TRPV2 is still an orphan receptor. The non-psychotropic phytocannabinoid, cannabidiol, was shown to activate TRPV2 expressed on sensory DRG neurons, resulting in CGRP release (Qin et al. 2008). Moreover, activation of TRPV2 by various physical stimuli induced degranulation of human mast cell line HMC1, which was inhibited by the nonselective TRPV2 blocker, SKF96365 (Zhang et al. 2012a). It was also suggested that the potentially pruritogenic PKA-dependent phosphorylation of TRPV2 can modulate channel activity in mast cells. However, TRPV2 as a potential target for preventing mast cell degranulation remains to be validated.
4.3 TRPV3 and TRPV4
TRPV3, a warm-sensitive channel, is highly expressed in keratinocytes (Nilius and Biro 2013; Nilius et al. 2013; Peier et al. 2002). In mice, a spontaneous gain-of-function (Gly573Ser) mutation in the trpv3 gene not only results in a hairless phenotype and dermatitis but also causes severe itching (Asakawa et al. 2006; Xiao et al. 2008). Transgenic mice overexpressing this mutant channel in their epidermal keratinocytes exhibit itching/scratching behavior and intracutaneous accumulation of NGF (Yoshioka et al. 2009). Importantly, Olmsted syndrome, a rare genetic disorder (TRP channelopathy) which is partly due to the same gain-of-function (Gly573Ser) TRPV3 mutation, is also characterized by debilitating itch (Lai-Cheong et al. 2012; Lin et al. 2012).
Somewhat unexpectedly, in an experimental dry skin model (evoked by application of acetone–ether–water), no differences were found in histological features of barrier disruption between wild-type and TRPV3-null animals, but the latter group showed less intensive scratching behavior attributed to decreased sprouting of cutaneous sensory C-fibers, suggesting a possible role of TRPV3 in dry skin-associated itch (Yamamoto-Kasai et al. 2012). TRPV3 can also regulate the release of various mediators like ATP, PGE2, or NO (Huang et al. 2008; Mandadi et al. 2009; Miyamoto et al. 2011), which can influence sensory functions, maybe involving itch.
Experimentally induced Mg2+ deficiency in mice or rats is a widely used model of AD-like scratching behavior (Chavaz et al. 1984; Neckermann et al. 2000). Intriguingly, both extra- and intracellular Mg2+ were shown to tonically inhibit TRPV3 activity in cultured epidermal keratinocytes (Luo et al. 2012). It was, therefore, speculated that hypomagnesemia (which can develop in various human disease conditions such as type 2 diabetes mellitus) may induce the overactivity of TRPV3, resulting in the development of pruritus. The role of TRPV3 in the development of AD is, however, controversial. The pathogenesis of AD is thought to involve compromised skin barrier formation, and the skin barrier is clearly defective in the trpv3 (−/−) animals. To explain the phenotype of the TRPV3-null mice, it was proposed that TRPV3 forms a complex with ADAM17, EGFR, and TGase1, which is required for normal terminal keratinocyte differentiation. Dysregulation of this complex (by absent TRPV3) may impair barrier function and lead to dry skin (Cheng et al. 2010b). Yet, as discussed above, TRPV3 KO mice showed less scratching than their wild-type littermates in an experimental AD model; conversely, the gain-of-function TRPV3 phenotype was associated with intense itching. To reconcile these seemingly contradictory findings, one might speculate that a certain level of basal TRPV3 activity is essential for normal skin function, with both increased and decreased activities leading to dermatological disease. Of note, disturbed barrier formation was detected in neonatal trpv3−/− mice, but not in adult TRPV3 KO animals with a different genetic background (Miyamoto et al. 2011; Moqrich et al. 2005).
TRPV4 as an itch target is highly controversial since none of the numerous human gain-of-function mutation channelopathies has a pruritic phenotype (Nilius and Voets 2013). Yet, in experimental models certain pruritogens were found to target TRPV4. For example, PAR-2 activation sensitizes TRPV4-coupled cellular responses in DRG neurons, resulting in increased neuropeptide release and mechanical hyperalgesia (Grant et al. 2007). Furthermore, in patients undergoing breast reduction surgery for painful mastopathy, increased expression of TRPV4 (as well as TRPV3) was detected in the basal layers of keratinocytes. This altered expression of TRPV4 was associated with an increase in TRPV1 and NGF expression in sensory fibers (Gopinath et al. 2005): this may intensify not only pain but also itch sensation. Furthermore, mast cell functions also seem to be affected by TRPV4. For instance, laser irradiation of mouse RBL-2H3 mast cells evoked intracellular Ca2+ increase and histamine release via TRPV4 (Yang et al. 2007). TRPV4 (like TRPV2) was also expressed by HMC1 human mast cells, and the increase of temperature to 37–39 °C, as well as the TRPV4 agonist 4α-phorbol 12,13-didecanoate, induced a weak outwardly rectifying current with subsequent increase in the intracellular Ca2+ concentration (Kim et al. 2010).
4.4 TRPA1
TRPA1 has emerged as a major target for histamine-independent itch. Chloroquine, an “itchy” antimalarial drug, was shown to activate both rodent (mouse and rat) MrgprA3 and human MrgprX1, members of the Mrgpr family that are exclusively expressed in peripheral sensory neurons (Dong et al. 2001). In mice, chloroquine-responsive, MrgprA3-expressing sensory neurons were activated by capsaicin, histamine, and BAM 8–22 (a specific MrgprC11 agonist), suggesting the co-expression of Mrgprs with TRPV1 and histamine receptors. Furthermore, these Mrgprs were also co-expressed with GRP, another neurotransmitter of the “itch pathway” (see above). Importantly, the deletion of the mrgpr gene cluster resulted in impaired chloroquine response, leaving the histamine-evoked scratching behavior intact: this observation implies a central role for Mrgprs in non-histaminergic itch (Liu et al. 2009).
TRPA1 is now thought to play a crucial role in the downstream signaling of both MrgprA3 and MrgprC11 (Wilson et al. 2011; Xiao and Patapoutian 2011). Indeed, in TRPA1 KO mice (in contrast to TRPV1 KO and wild-type animals) the activation of neither MrgprA3 (by chloroquine) nor MrgprC11 (by BAM 8–22) resulted in itch-related scratching behavior. Although in wild-type animals these substances evoked an elevation in the intracellular Ca2+ concentration and a resultant firing of action potentials in the capsaicin- and mustard oil-sensitive subsets of DRG neurons, respectively, these effects were abolished in the TRPA1 KO mice (but not in the TRPV1 KO animals). Furthermore, in heterologous expression systems, activation of the Mrgprs was able to evoke Ca2+ responses only in cells in which they were co-expressed with TRPA1. Finally, pharmacological experiments suggested that MrgprA3 was coupled to TRPA1 via the βγ subunit of G proteins, whereas MrgprC11 most probably signals via a PLC-dependent pathway (Wilson et al. 2011).
The situation is, however, probably even more complex. A recent study demonstrated that only a slight minority (~43 %) of chloroquine-sensitive DRG neurons respond to the TRPA1 agonist mustard oil, whereas the majority of the mustard oil-insensitive cells were activated by the TRPV1-specific agonist, capsaicin. Intriguingly, a minor population responded to neither TRPA1 nor TRPV1 stimulation (Than et al. 2013). Unexpectedly, these TRPA1-negative neurons seem to respond to chloroquine in a TRPC3-mediated fashion. If confirmed, this observation will furnish TRPC3 with a previously unexpected function in itch pathogenesis.
There is good evidence that TRPA1 is also involved in other pruritogenic mechanisms. For example, hydrogen peroxide-induced itch is clearly mediated by TRPA1, as it is inhibited by the pharmacological blockade of TRPA1 but not by TRPV1. Moreover, the hydrogen peroxide-induced itch was not influenced by histamine receptor blockers (Liu and Ji 2012). By contrast, scratching behavior induced by intradermal LTB4 administration was attenuated by the TRPA1 antagonist TCS-5861528 and the TRPV1 antagonist SB366791 with comparable efficacies (82 % inhibition and 97 % inhibition, respectively), but their mechanisms likely differ. TRPA1 was found to play a role in increased superoxide production in contrast to TRPV1, the deletion of which resulted in impaired myeloperoxidase release (Fernandes et al. 2013). In addition, TRPA1 might play a role in mediating the activation of sensory neurons by PAR-2, which was shown to sensitize TRPA1 by most likely degrading PIP2 by PLC (Dai et al. 2007). Further, TRPA1 (apparently in concert with TRPV1) seems to be involved in the downstream signaling of itch, generated by the activation of IL-31 receptor (IL-31RA) on DRG sensory neurons (Cevikbas et al. 2013). IL-31-evoked responses were significantly reduced both in TRPA1 and TRPV1 knockout mice. Most recently, TRPA1 was also found to be involved in the itch development in AD. In a mouse model, the thymic stromal lymphopoietin (TSLP) produced by atopic keratinocytes excited sensory neurons via the TSLP receptor–PLC–TRPA1 pathway and resulted in intense scratching behavior. The TSLP production of keratinocytes was stimulated by PAR-2 activation (Wilson et al. 2013).
Unexpectedly, it was reported that TRPA1 blockade enhanced ET-1 (but not histamine)-induced itch responses (Liang et al. 2011), suggesting a paradoxical inhibitory role of TRPA1 in (at least some forms of) histamine-independent itch. This controversy indicates the urgent need for further studies to clarify the contribution of TRPA1 to other forms of itch.
Although TRPA1 expression is considered to be remarkably selective for neurons, TRPA1 expression was also described in non-neuronal cells. Indeed, TRPA1 expressed in lung fibroblasts may play an important role in nonneurogenic inflammatory responses in diseased airways (Nassini et al. 2012). Pertinent to the topic of this review, TRPA1 expression was reported in epidermal keratinocytes. Exposure of NHEKs to low temperatures (13–15 °C) or TRPA1 agonists (allyl isothiocyanate or cinnamaldehyde) induced elevation of [Ca2+]e, which was prevented by the TRPA1 antagonist, HC-030031. Interestingly, these effects were more pronounced in undifferentiated cells (Tsutsumi et al. 2010). Moreover, treatment of NHEKs with icilin (activator of both TRPA1 and TRPM8, another cold-sensitive channel) caused alterations in the expressions of adhesion and extracellular matrix components, as well as molecules regulating cell cycle, apoptosis, and differentiation (Atoyan et al. 2009; commented in Biro and Kovacs 2009). Taken together, these observations suggest that TRPA1 in keratinocytes may be involved in the regulation of the epidermal barrier. Indeed, in mice following tape striping, topical application of TRPA1 agonists accelerated barrier recovery, and this effect was prevented by pretreatment with HC-030031. Interestingly, HC-030031 alone delayed the barrier recovery which argues for a “constitutive” (tonically active) role of TRPA1 in epidermal barrier homeostasis. Local cooling of the skin (10–15 °C for 1 min) evoked similar effects, most probably via accelerated secretion of (barrier-forming) lamellar bodies at the interface of stratum granulosum and corneum; this action was also inhibited by the TRPA1 antagonist (Denda et al. 2010b).
Stimulation of TRPA1 expressed on NHEKs also induced the synthesis of the proinflammatory interleukins, IL-1α and IL-1β (Atoyan et al. 2009). Moreover, topical application of the TRPA1 agonist, cinnamaldehyde, induced skin inflammation. Interestingly, although the edema component was prevented by aprepitant (an antagonist of the tachykinin NK1 receptor), it was not affected by HC-030031. By contrast, the cinnamaldehyde-induced leukocyte infiltration was effectively suppressed by pharmacological TRPA1 blockade, while the NK1 antagonist was ineffective (Silva et al. 2011). In a mouse contact hypersensitivity model, TRPA1 activation enhanced ear swelling response and migration of dendritic cells to draining lymph nodes, which the effect was antagonized by HC-030031 (Shiba et al. 2012).
A recent study testing various agents known to induce contact dermatitis in humans verified the central role of TRPA1 activation and resultant SP release in the development of contact dermatitis. Genetic deletion or pharmacological blockade of TRPA1 (but not TRPV1) resulted in decreased skin edema, keratinocyte hyperplasia, leukocyte infiltration, and scratching behavior in mice exposed to oxazolone. In oxazolone-challenged skin of TRPA1-deficient mice, a significant decrease was found in the expression of inflammatory cytokines, NGF, and endogenous pruritogens (e.g., SP and serotonin). Oxazolone was also shown to activate recombinant TRPA1, further supporting the causal role of the channel in contact dermatitis. Urushiol, the contact allergen in poison ivy, evoked similar responses which were diminished in TRPA1-null mice (Liu et al. 2013).
4.5 TRPM4
TRPM4 is a Ca2+-activated, nonselective cation channel which negatively regulates the driving force of Ca2+ ion influx in mast cells. Indeed, bone marrow-derived mast cells obtained from TRPM4 KO animals showed increased degranulation and mediator release in vitro, as well as a more severe IgE-mediated acute passive cutaneous anaphylactic response in vivo (Vennekens et al. 2007). Genetic deletion of TRPM4 dysregulated the migration of mast cells (Shimizu et al. 2009). Taken together, these observations imply a therapeutic potential for TRPM4 agonists in the management of anaphylactic reactions or local allergic responses. Unfortunately, the widespread expression of TRPM4 and its involvement in various physiological functions (most important, cardiac impulse generation) may strongly limit (or completely prevent) the systemic application of TRPM4 activators in clinical practice.
4.6 TRPM6 and TRPM7
In rats, insufficient dietary Mg2+ intake leads to low serum Mg2+ concentrations (hypomagnesemia) and subsequent development of dermatitis and intense scratching behavior (Thomsen et al. 2005). Indeed, uremic patients on hemodialysis with low serum Mg2+ levels develop itch which disappears after the concentration of Mg2+ in the dialysate is normalized (Graf et al. 1979). The emerging link between low Mg2+ and itch has brought TRP channels (TRPM6 and TRPM7) that play a role in Mg2+ homeostasis (the so-called Mg2+nificent TRP channels) in the center of spotlight.
Epidermal growth factor (EGF), a central growth factor in epidermal differentiation, has a crucial role in controlling TRPM6 functions (Muallem and Moe 2007). Indeed, EGF stimulates Mg2+ reabsorption in the renal distal convoluted tubule. A point mutation in pro-EGF was shown to disturb this mechanism, resulting in renal Mg2+ loss (Groenestege et al. 2007). In support of a link among EGF, TRPM6, Mg2+, and itch, several studies described itch as a common side effect of cetuximab, a chemotherapeutic monoclonal antibody that inhibits the EGF receptor (Agero et al. 2006; Porzio et al. 2006; Wu et al. 2011).
4.7 TRPM8
Menthol, the classical activator of TRPM8, is an active ingredient in various OTC topical anti-itch lotions. Indeed, several case studies reported a modest clinical benefit for menthol in various forms of pruritus (Frolich et al. 2009; Haught et al. 2008). For example, menthol (as well as causing moderate cooling of the skin) decreased the subjective intensity of histamine-induced itch (Bromm et al. 1995). Furthermore, menthol provided relief in mustard gas-induced pruritus in chemical warfare-injured veterans (Panahi et al. 2007). Controlled clinical trials, however, found no evidence that menthol is more effective than placebo in relieving pruritus. Indeed, menthol was not only ineffective to suppress histamine-induced itch, but, even worse, it increased the transepithelial water loss, suggesting an irritant nature of the compound (Yosipovitch et al. 1996).
Although TRPM8 is often referred to as the menthol receptor, menthol is not selective for TRPM8. For example, menthol was reported to activate TRPV3 (Macpherson et al. 2006) and to either inhibit or activate TRPA1 in a dose- and species-dependent manner (Karashima et al. 2007; Xiao et al. 2008).
TRPM8 appears not to be involved in the development and/or exacerbation of itch sensation, although it may exert an inhibitory role. Indeed, pruritogenic proinflammatory mediators (such as bradykinin and PGE2) desensitize the cooling-evoked and most likely TRPM8-mediated calcium transients and shift the activation threshold toward colder temperatures (Linte et al. 2007). This inhibitory effect of the proinflammatory mediators was proposed to target TRPM8 via a quite “unusual” signaling mechanism, an α-subunit of a G protein, directly bound to TRPM8 (Zhang et al. 2012b).
Our understanding of the roles that non-neuronal TRPM8 may play in itch sensation is both limited and controversial. For example, TRPM8 expression was described in the RBL-2H3 basophilic leukemia mast cell line. In these cells, activation of TRPM8 by menthol and cold stimuli evoked an elevation in the intracellular Ca2+ concentration and induced histamine release. These effects were blocked both pharmacologically and by siRNA-based silencing of TRPM8 (Cho et al. 2010). By contrast, another study could not detect any TRPM8 expression in human mast cells. Moreover, TRPM8 did not appear to play any role in mast cell degranulation (Medic et al. 2011). Recently, TRPM8 expression (both at mRNA and protein levels) was detected in epidermal keratinocytes isolated from hairless mice. In these animals, following epidermal barrier disruption by tape stripping, topical application of menthol or the TRPM8 agonist WS12 potentiated the barrier recovery; this effect was blocked by the general TRP antagonist ruthenium red, as well as the nonselective TRPM8 antagonist, BCTC (Denda et al. 2010a). Chloroquine (a pruritogenic activator of TRPA1 and maybe also TRPC3) was reported to inhibit TRPM8 indirectly via cellular signaling pathways (Than et al. 2013). Last, Han et al. (Han et al. 2012) reported a beneficial effect for the topical TRPM8 agonist icilin in vulvar pruritus secondary to Lichen sclerosus et atrophicus.
4.8 Canonical TRP Channels
Calcium signaling is a key mechanism in keratinocyte differentiation (Bikle et al. 2001; Micallef et al. 2009; Tu et al. 2004; Yuspa et al. 1988), and altered differentiation of epidermal keratinocytes may play a role in pruritic diseases such as AD and psoriasis (Hanifin 2009). TRPC channels are important regulators of intracellular calcium homeostasis in several cell types (Clapham 2003; Nilius and Mahieu 2006; Nilius and Voets 2005; Ramsey et al. 2006; Talavera et al. 2008), including epidermal or mucosal keratinocytes. The expression level of individual TRPC channels (TRPC1, TRPC4, TRPC5, TRPC6, and TRPC7) in keratinocytes fluctuates in a differentiation-dependent manner (Cai et al. 2005, 2006; Fatherazi et al. 2007).
TRPC1 is overexpressed in the epidermis of patients with Darier’s disease (DD), a genetic disorder caused by loss-of-function mutations in the SERCA2b gene that encodes the Ca2+ pump of the endoplasmic reticulum. This causes a severe differentiation disorder of keratinocytes which is often associated with intense pruritus (Barfield et al. 2002; Pani et al. 2006). In keratinocytes obtained from DD patients, TRPC1-mediated Ca2+ influx was significantly higher compared to healthy subjects. Furthermore, DD keratinocytes show enhanced proliferation and apoptosis resistance, suggesting that TRPC1 is involved in the abnormal keratinization in DD epidermis. Importantly, experiments performed in SERCA2b KO mice, as well as on human epidermal HaCaT keratinocytes in which the expression of SERCA2b was silenced by siRNA, yielded similar results (Pani et al. 2006). Other studies showed that TRPC1/TRPC4 heteromers were important for keratinocyte differentiation as siRNA-based silencing of these channels prevented the induction of Ca2+-induced differentiation. In basal cell carcinoma (BCC), the lack of TRPC1/TRPC4 heteromers was coupled to impaired differentiation and enhanced proliferation (Beck et al. 2008).
Furthermore, the activation of TRPC6 expressed in both HaCaT and NHEKs induced differentiation and inhibited proliferation by increasing Ca2+ influx (Leuner et al. 2011; Muller et al. 2008; Woelfle et al. 2010). In addition, decreased expression of TRPC1/TRPC3/TRPC4/TRPC5/TRPC6 and TRPC7 was found in keratinocytes obtained from psoriasis patients, another pruritic dermatosis characterized by a disturbed proliferation–differentiation program (Leuner et al. 2011)]. Most recently, TRPC6 has been implicated in the pathogenesis of AD (Sun et al. 2012). Collectively, these data argue for a role of certain TRPCs in itch development. Indeed, as discussed above, TRPC3 was implicated in the chloroquine-induced activation of TRPA1-negative neurons (Than et al. 2013).
5 Targeting TRP Channels for Itch Relief: The Promise and the Challenges
Without doubt, TRP channels (and drugs that target them) have become hot topics in biomedical research during the past three decades as demonstrated by the almost exponential growth in the number of papers in scientific publications (Kaneko and Szallasi 2013). The most studied TRP channel is no doubt TRPV1. Indeed, a number of potent small molecule TRPV1 antagonists have advanced to clinical trials for indications of pain (also chronic cough and overactive bladder), but a combination of unforeseen adverse effects (most important, hyperthermia and impaired heat pain sensation) and disappointing clinical efficacy has prevented any compounds from progressing beyond Phase II (Brederson et al. 2013; Moran et al. 2011). These disappointing results emphasize a number of questions that already emerged after the failure of attempts to translate the compelling basic research findings into clinical applications. Most important: do currently used pain (itch) models accurately reflect the biological situation in patients suffering from chronic pain (itch)? For example, TRPV1 antagonist block evoked cough (e.g., inhaled citric acid) in study volunteers but are without any clinical benefit in chronic cough patients (Kaneko and Szallasi 2013). It is with these questions in mind that we have to ponder the clinical promise of TRP channel-targeting drugs as novel antipruritic agents.
Chronic itch is a large, unmet medical need. According to a questionnaire-based German study, the lifetime prevalence of chronic itch exceeds 20 % (Matterne et al. 2013). In animal experiments, TRPV1 has been linked to histamine-induced itch, and TRPA1 has emerged as a primary target for chloroquine and urushiol, the contact allergen in poison ivy. TRPV1 and TRPM6/TRPM7 have also been implicated in the pathogenesis of pruritus that develops in patients with kidney (uremia) or liver failure. A gain-of-function TRPV3 phenotype was associated with itch both in experimental animals and in patients (Olmsted syndrome). Last, enhanced TRP channel (e.g., TRPV1, TRPV3, and various TRPCs) expression was described in various pruritic skin disorders. Taken together, these findings imply a clinical potential for targeting TRP channels in patients with pruritus.
However, several outstanding questions about the following remain, including:
Identification of physiological roles of TRP channels expressed on non-neuronal cells of human skin during inflammation and its related production of pruritogens
Definition of relationship of TRP channels to peripheral and central itch processing (sensitization, desensitization)
Determination of the expression patterns of individual TRP channels in various pruritic dermatoses
Investigation of prurigo nodularis-like lesions in TRP channel gene-deficient mice
In vivo evaluation of “combination therapies,” using substances that target different TRP channels
Synthesis of selective drugs targeting TRP channels and their evaluation in vitro as well as in vivo
In conclusion, there is no uniformly effective pharmacological intervention to treat all pruritic diseases because no single target/mediator is responsible for itch. Clearly, we need to accept the need for a combination of drugs. During the past few years, tremendous progress has been made in our understanding of the roles that TRP channels play in itch sensation in preclinical models. Selective and effective TRP channel blockers are indispensible to corroborate the relevance of these compelling basic research findings in the pharmacotherapy of pruritus in patients. For some channels (e.g., TRPV1, TRPV3, and TRPA1) such antagonists are already available. For others (e.g., TRPC3, TRPM4, and TRPM6/TRPM7) the development of potent and selective agonists/antagonists is still awaited. It remains to be seen if TRP channel drugs will become components of a clinically effective itch therapy regimen.
Acknowledgments
The writing of this review was supported by Hungarian (“Lendület” LP2011-003/2011, OTKA 101761, OTKA 105369, TÁMOP-4.2.2./A-11/1/KONV-2012-0025) and EU (FP7-REGPOT-2008-1/229920) research grants as well as funding from the People Programme (Marie Curie Actions) of the European Union’s Seventh Framework Programme (FP7/2007–2013) under REA grant agreement n° 330489.
References
Abadia Molina F, Burrows NP, Jones RR, Terenghi G, Polak JM (1992) Increased sensory neuropeptides in nodular prurigo: a quantitative immunohistochemical analysis. Br J Dermatol 127:344–351PubMed
Agero AL, Dusza SW, Benvenuto-Andrade C, Busam KJ, Myskowski P, Halpern AC (2006) Dermatologic side effects associated with the epidermal growth factor receptor inhibitors. J Am Acad Dermatol 55:657–670PubMed
Akiyama T, Carstens E (2013) Neural processing of itch. Neuroscience 250:697–714PubMedCentralPubMed
Akiyama T, Carstens MI, Carstens E (2010) Enhanced scratching evoked by PAR-2 agonist and 5-HT but not histamine in a mouse model of chronic dry skin itch. Pain 151:378–383PubMedCentralPubMed
Akiyama T, Iodi Carstens M, Carstens E (2011) Transmitters and pathways mediating inhibition of spinal itch-signaling neurons by scratching and other counterstimuli. PLoS One 6:e22665PubMedCentralPubMed
Amadesi S, Nie J, Vergnolle N, Cottrell GS, Grady EF, Trevisani M, Manni C, Geppetti P, McRoberts JA, Ennes H, Davis JB, Mayer EA, Bunnett NW (2004) Protease-activated receptor 2 sensitizes the capsaicin receptor transient receptor potential vanilloid receptor 1 to induce hyperalgesia. J Neurosci 24:4300–4312PubMed
Amantini C, Mosca M, Lucciarini R, Perfumi M, Morrone S, Piccoli M, Santoni G (2004) Distinct thymocyte subsets express the vanilloid receptor VR1 that mediates capsaicin-induced apoptotic cell death. Cell Death Differ 11:1342–1356PubMed
Amaya F, Shimosato G, Nagano M, Ueda M, Hashimoto S, Tanaka Y, Suzuki H, Tanaka M (2004) NGF and GDNF differentially regulate TRPV1 expression that contributes to development of inflammatory thermal hyperalgesia. Eur J Neurosci 20:2303–2310PubMed
Andoh T, Kuraishi Y (1998) Intradermal leukotriene B4, but not prostaglandin E2, induces itch-associated responses in mice. Eur J Pharmacol 353:93–96PubMed
Andoh T, Katsube N, Maruyama M, Kuraishi Y (2001) Involvement of leukotriene B(4) in substance P-induced itch-associated response in mice. J Invest Dermatol 117:1621–1626PubMed
Andoh T, Nishikawa Y, Yamaguchi-Miyamoto T, Nojima H, Narumiya S, Kuraishi Y (2007) Thromboxane A2 induces itch-associated responses through TP receptors in the skin in mice. J Invest Dermatol 127:2042–2047PubMed
Andrew D, Craig AD (2001) Spinothalamic lamina I neurons selectively sensitive to histamine: a central neural pathway for itch. Nat Neurosci 4:72–77PubMed
Ansel JC, Armstrong CA, Song I, Quinlan KL, Olerud JE, Caughman SW, Bunnett NW (1997) Interactions of the skin and nervous system. J Investig Dermatol Symp Proc 2(1):23–26PubMed
Arai I, Tsuji M, Takeda H, Akiyama N, Saito S (2013) A single dose of interleukin-31 (IL-31) causes continuous itch-associated scratching behaviour in mice. Exp Dermatol 22:669–671PubMed
Arrang JM, Garbarg M, Schwartz JC (1983) Auto-inhibition of brain histamine release mediated by a novel class (H3) of histamine receptor. Nature 302:832–837PubMed
Asakawa M, Yoshioka T, Matsutani T, Hikita I, Suzuki M, Oshima I, Tsukahara K, Arimura A, Horikawa T, Hirasawa T, Sakata T (2006) Association of a mutation in TRPV3 with defective hair growth in rodents. J Invest Dermatol 126:2664–2672PubMed
Atoyan R, Shander D, Botchkareva NV (2009) Non-neuronal expression of transient receptor potential type A1 (TRPA1) in human skin. J Invest Dermatol 129:2312–2315PubMed
Averbeck B, Reeh PW (2001) Interactions of inflammatory mediators stimulating release of calcitonin gene-related peptide, substance P and prostaglandin E(2) from isolated rat skin. Neuropharmacology 40:416–423PubMed
Axelsson HE, Minde JK, Sonesson A, Toolanen G, Hogestatt ED, Zygmunt PM (2009) Transient receptor potential vanilloid 1, vanilloid 2 and melastatin 8 immunoreactive nerve fibers in human skin from individuals with and without Norrbottnian congenital insensitivity to pain. Neuroscience 162:1322–1332PubMed
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


