and Marc Tramier1
(1)
Institut de Génétique et Développement de Rennes, UMR 6290, CNRS, Université de Rennes 1, Rennes, France
(2)
Institut Jacques Monod, UMR 7592, CNRS, Université Paris-Diderot, Paris, France
Abstract
Dual-color FCS is a powerful method to monitor protein–protein interactions in living cells. The main idea is based on the cross-correlation analysis of temporal fluorescence intensity fluctuations of two fluorescent proteins to obtain their co-diffusion and relative concentration. But, when performing these experiments, the spectral overlap in the emission of the two colors produces an artifact that corrupts the cross-correlation data: spectral bleed-through. We have shown that problems with cross talk are overcome with Fluorescence Lifetime Correlation Spectroscopy (FLCS). FLCS applied to dual-color cross-correlation, utilizing for example eGFP and mCherry fluorescent proteins, allows the determination of protein–protein interactions in living cells without the need of spectral bleed-through calibration. Here, we present in detail how this methodology can be implemented using a commercial setup (Microtime from PicoQuant, SP8 SMD from Leica or any conventional confocal with PicoQuant TCSPC module, and also with a Becker and Hickl TCSPC module). The dual-color FLCS experimental procedure where the different laser intensities do not have to be controlled during the experiment constitutes a very powerful technique to quantitatively study protein interactions in live samples.
Key words
FCSFCCSFLCSQuantitative fluorescence microscopyTCSPC1 Introduction
The study of the dynamics of macromolecular complexes becomes essential to investigate molecular mechanisms in live cells. Different approaches are used to investigate protein–protein interactions in live cells. To this end, we are here interested in dual-color fluorescence correlation spectroscopy (FCS) [1]. The technique consists on the cross-correlation analysis of temporal fluorescence intensity fluctuations of two color fluorophores to follow the co-diffusion of two proteins labeled with these fluorescent tags. Dual-color FCS is performed at a single point and is capable of detecting interactions between proteins in which FRET does not necessarily occur, therefore provides complementary information relative to FRET in imaging mode. This approach overcomes the main limitation of FRET that requires the close proximity of the two fluorescent tags. The only requirement in order to detect interactions using dual-color FLCS is that both fluorescent proteins should diffuse and/or transiently interact with high molecular weight complexes or mobile subcellular structures. Recently, this approach was used in live cells giving novel insights on the study of macromolecular complexes, such as the MAP kinase complexes in yeast pheromone signaling [2], the dimerization of EGF receptors [3], the T cell receptor complexes [4], and the virus RNA polymerase assembly [5]. But couples of protein tags commonly used in live cells (ECFP/EYFP and EGFP/mCherry) present spectral overlap in their emission giving rise to spectral bleed-through that corrupts the cross-correlation data [5]. The cross-talk contribution from the shorter wavelengths emission species in the longer wavelengths channel is usually minimized by choosing red-shifted band-pass filter for the longer wavelengths channel and/or by decreasing the intensity of the laser exciting the shorter wavelengths emission species. But these tricks decrease the brightness of both fluorescent species, a parameter usually critical in dual-color FCS in live cells. The cross-talk can also be calibrated and its amount retrieved during data analysis [6]. But nonetheless it impedes the study of very low amounts of protein–protein interactions since at least 10 % of cross talk is always observed when working with green and red fluorescent proteins. The use of novel fluorescent proteins exhibiting a large Stokes shift in dual-color FCS experiments decreases the contribution of the spectral cross talk to the cross-correlation [7, 8], but the weak photo stability of some of them introduces other difficulties.
To eliminate spectral bleed-through in dual-color FCS, the switching method or pulsed interleaved excitation was introduced [9–11]. In this method, two alternating laser beams are controlled by an acousto-optic modulator and the synchronized correlation signals in both detectors are calculated as cross-correlation functions with no cross-talk. These approaches necessitate homemade developed confocal setups and cannot be easily implemented to commercial microscopes. A similar method was recently implemented in dual-color scanning FCS with a conventional confocal microscope to quantify ligand-receptor interactions in cell membranes of living zebrafish embryos [12]. In membranes, the diffusion of protein complexes is slow, which can be monitored by using the sequential dual-color scanning method. However, the speed of acquisition of line-scanning FCS in sequential dual color is too slow to investigate intracellular co-diffusion.
Here, we propose a FCCS-based method to overcome the EGFP and mCherry spectral cross talk in quantitative studies of protein–protein interactions in live cells by using a commercial confocal system (Microtime from PicoQuant, SP8 SMD from Leica or any conventional confocal TCSPC module) (e.g., PicoQuant or Becker & Hickl). Our approach is based on Fluorescence Lifetime Correlation Spectroscopy (FLCS). The use of a pulsed laser to perform FCS takes advantage of Time Correlated Single Photon Counting (TCSPC) to differentiate the contribution of two different fluorescence lifetime species to FCS file (FLCS, [13]). Indeed, each single photon is recorded with its global arrival time and the delay time with respect to the corresponding laser pulse. The FLCS method relies on the calculation of lifetime filters for each species having different fluorescence decay signatures in the TCSPC histogram. Each fluorescent component, in the fluorescent time trace, having different lifetime properties can be resolved on the basis of its fluorescence decay signature. FLCS was already used in spectroscopy to suppress scattered light and detector afterpulses [14], to study dynamic transition between two fluorescence lifetime states [15], to resolve different lifetime species [16], and to measure precise concentration values as low as picomolar of fluorophore [17]. FLCS was also employed in live cells to quantify the interaction of EGFR-neutralizing antibody labeled with Alexa488 and EGFR-GFP by fluorescence lifetime cross-correlation measurements [18]. Recently, we have shown that FLCS applied to dual-color EGFP and mCherry cross-correlation allows the determination of protein–protein interactions in living cells without any spectral bleed-through artifact [19].
By using two lasers to excite simultaneously two spectrally different fluorophores, a pulsed laser (470 nm which can excite a green fluorescent species, i.e., EGFP) and a continuous laser (561 nm to excite a red fluorescent protein, i.e., mCherry), dual-color FLCS provides a way to remove the spectral bleed-through of the green species into the red detection channel. The photons coming from the green species detected in the red channel are time correlated to the pulsed laser (see the fluorescence decay histogram in Fig. 1a, top) in contrast to the photons coming from the red species which do not harbor any fluorescence decay signature (see the constant emission histogram in Fig. 1a, middle). When both fluorophores are excited together, the fluorescence histogram contains the two signals (fluorescence decay for the spectral bleed-through of the green species and constant histogram for the red species, Fig. 1a, bottom). Lifetime filter functions are calculated from the two-component fluorescence decay analysis acquired in the red channel. For each time-resolved channel, the lifetime filter value gives the statistical weight of the counted photon for its corresponding species. In Fig. 1b are shown the filter corresponding to the fluorescence decay species (in blue) and this corresponding to the steady-state fluorescence (in pink). These different filters can be selected during auto- and cross-correlation process of the absolute time traces to take into account the different weighting of each individual photon. By using the lifetime filter of the fluorescence decay of EGFP in the green channel and the filter of the steady-state fluorescence of mCherry in the red channel during cross-correlation calculation of the green and red time trace, the spectral bleed-through artifact is completely removed. We stress the potentiality of the technique when applied to quantify protein–protein interactions in vivo where protein A is fused to EGFP and protein B is fused to mCherry. The cross-correlation amplitude is directly dependent on the quantity of protein complexes and is not related to both the relative concentration of green and red species and the relative laser intensities used during the experiment. The dual-color FLCS experimental procedure where the different laser intensities do not have to be controlled during the experiment is extremely powerful to quantitatively study protein interactions in live samples.
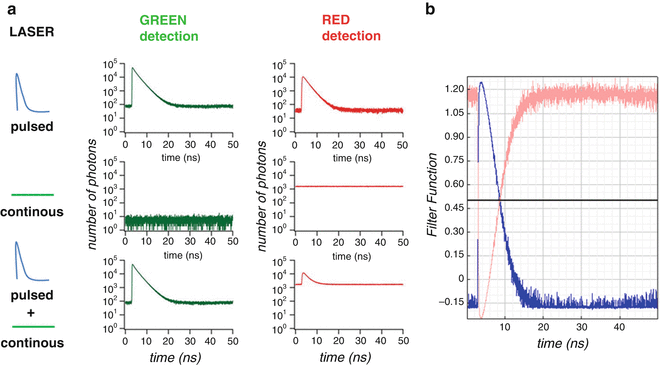
Fig. 1
Dual-color FLCS approach and fluorescence lifetime filters. (a) Fluorescence time-correlation histograms of GFP and mCherry fluorescence detected in the green (left) and red (right) channels by using 470 nm pulsed laser alone (upper), 561 nm CW laser (middle), and both (lower). (b) Fluorescence lifetime filters of the fluorescence decay (blue) and the steady-state fluorescence (pink) calculated by Symphotime software (PicoQuant) for the fluorescence decay histogram of both GFP and mCherry in the red channel detection by using 470 nm pulsed and 561 nm CW laser excitation (red signal in a)
2 Materials
2.1 Live Cells Sample Preparation
Dual-color FLCS has been optimized for green and red fluorescent proteins. In this protocol, we present the technique to be employed to detect very low amounts of interacting proteins in He-La cells (i.e., A-GFP + B-mCherry where A and B are the proteins of interest). The method can be adapted to any other cell line or other live sample expressing the two proteins, with the limitation of having sufficiently low amount of fluorescent proteins for a FCS experiment (see Note 1).
1.
Culture He-La cells in Dulbecco’s modified eagle medium containing 10 % fetal bovine serum (PAA Laboratories GmbH, Pasching, Austria) at 37 °C in a humidified atmosphere of 5 % CO2.
2.
Seed the cells on 32-mm round glass coverslip at a density of 2 × 105 cells. When cells are around 50–70 % of confluence, perform a transient transfection using the proteins A-GFP and B-mCherry. Also, perform a second transfection using EGFP and mCherry expressing plasmids and mCherry-EGFP tandem expressing plasmid with a total amount of 1 μg of expression vectors (see Note 2) using Nanofectin I (PAA).
3.
24 h after transfection, mount the cells in an open observation chamber with special medium without phenol red (we use special DMEM-F12 without phenol red, B12 vitamin, riboflavin and supplemented with 20 mM Hepes, and l-glutamine from PAA) and with low amount of serum (with He-La cells no serum is required) to prevent autofluorescence from the sample. Add 1 ml of parafilm oil (Merck) to avoid evaporation at 37 °C.
4.
Use a microscope incubation chamber (for example PeCon GmbH or LIS) to perform live cell measurements at 37 °C, if possible with 5 % CO2 flux.
2.2 Microscopy Setup
A confocal light scanning microscope equipped with pulsed laser excitation, electronics to carry out Time Correlated Single Photon Counting (TCSPC), and single photon counting detectors (i.e., SPADS and/or APD) is needed to perform FLCS measurements. We carried out all measurements presented here employing the PicoQuant Microtime 200 [16, 17, 19] but other solutions are available in the market, for instance, the SP8/SP5 combined with the SMD module by Leica (Manheim, Germany) or the combination of any confocal with a PicoQuant TCSPC module. In the rest of the protocol, we describe how to carry dual-color FLCS with a Microtime 200, but the reader should be able to translate this description to his/her own system. Figure 2 shows the optical pathway. Two laser excitation sources (if we are to use GFP and mCherry, a pulsed blue laser operating at 470 nm and a continuous green laser operating at 561 nm) are directed into a water objective with a high numerical aperture objective (60×, NA 1.3) mounted in an inverted microscope and is directly focused onto co-transfected live cells. The fluorescence emission is focused through a pinhole and the corresponding emission filters (510DF40, 580DF20) and is collected by two avalanche photodiode detectors (SPAD 1 and SPAD 2). The PicoQuant TimeHarp 300 module is employed in a Time-Tagged Time-Resolved (TTTR) mode (see Note 3) and is coupled to a PC that pilots the data acquisition and analysis using the PicoQuant Symphotime software:
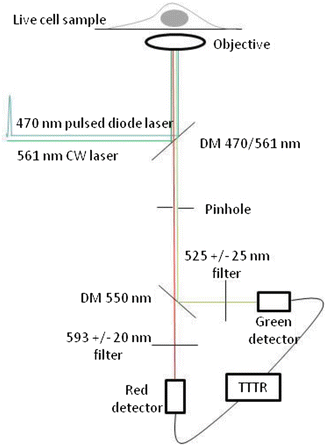
Fig. 2
Dual-color FLCS microscope. Two lasers (470 nm pulsed and 561 nm CW) are conducted into epi-fluorescence microscope for live sample measurement. The confocal fluorescence selected by a pinhole is split into green and red channels. Single photon counting detectors (SPADs) are used in Time-Tagged Time-Resolved (TTTR) mode
1.
Select the pulsed diode tuned at 470 nm (PicoQuant) to excite EGFP and a continuous DPSS laser at 561 nm (Coherent) to excite mCherry.
2.
Utilize a water immersion objective with a high numerical aperture (for instance, we recommend a 60×, NA 1.3) mounted on an inverted microscope.
3.
Select a double dichroic mirror (DM 470/571 nm) and adjust the pinhole aperture for the two colors (see Note 4).
4.
Select the right emission filters for GFP and mCherry (525 ± 25 for the green channel and 593 ± 20 for the red channel) (see Note 5).
5.
Utilize as detection two avalanche photodiodes (for instance, SPAD from Perkin Elmer, Tau-SPAD or MPD SPAD from PicoQuant, see Note 6).
3 Methods
We present here the protocol for dual-color FLCS in the context of A-GFP and B-mCherry co-diffusion study to reveal protein–protein interactions. We suppose that the system is already calibrated in terms of conventional FCS (calibration of the confocal volume, see Note 7). We also encourage readers using GFP and mCherry alone and in tandem expressed in cells to validate the technique with their own system ([19], see Note 8).
3.1 Dual-Color FLCS in Live Cells
1.
Choose, using the mercury lamp set at low intensities, the right cell in which to park the confocal beam. Picking up the right cells (low expressing levels of fluorophore) is essential in order to get the right data (see Note 9).
2.
Park the beam in different areas of the cell and check the intensity trace (it should not be higher than 200 kHz) by adjusting the different laser intensities. The correction of the spectral bleed-through by FLCS is not related to the laser intensities. For example, try diminishing the power in the green laser (561 nm excitation) only, this will have a drastic effect in false cross-correlation (bleed-through coming from green photons detected in the red channel). This is only true with conventional dual-color FCS approaches, but not in dual-color FLCS. This is crucial to investigate very low amounts of proteins in interaction and makes FLCS a unique technique in terms of sensitivity. To increase the laser power in this scenario for both channels, but especially for the mCherry channel, is crucial, since the relatively low sensitivity of the SPADS in the red part of the spectrum and the lower photostability of mCherry compared to GFP require particular intensity conditions that may change from cell to cell. This is why the independence of FLCS to intensity (as is the case in FLIM) constitutes a great improvement compared to FCCS.
3.
Check that the shape of the intensity trace is steady over time (indicating that your measurement is not affected by photobleaching) in both channels. Decrease the laser intensities if photobleaching occurs.
4.




Acquire the whole data for about 30 s to 1 min in a TTTR file which contains all the single photon events with their respective absolute time, correlated time, and detection channels. Using Symphotime software, it is easy to play TTTR file to recover the different histograms: fluorescence decays of both channels and time trace of both channels. Symphotime is also designed to calculate auto- and cross-correlations of the different time traces, particularly using calculated lifetime filter from the fluorescence decays (see below).
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
