Chapter 20 Psychotropic drugs
Diagnostic issues
Also falling within the scope of modern psychiatric diagnostic systems are organic mental disorders (e.g. dementia in Alzheimer’s disease), disorders due to substance misuse (e.g. alcohol and opiate dependence; see Ch. 11), personality disorders, disorders of childhood and adolescence (e.g. attention deficit/hyperactivity disorder, Tourette’s syndrome), and mental retardation (learning disabilities).
Antidepressant drugs
Antidepressants can be broadly divided into four main classes (Table 20.1), tricyclics (TCA, named after their three-ringed structure), selective serotonin reuptake inhibitors (SSRIs), monoamine oxidase inhibitors (MAOIs) and novel compounds, some of which are related to TCAs or SSRIs. Clinicians who wish to have a working knowledge of antidepressants would be advised to be familiar with the use of at least one drug from each of the four main categories tabulated. A more thorough knowledge-base would demand awareness of the distinct characteristics of the subgroups of novel compounds (e.g. serotonin and noradrenaline/norepinephrine reuptake inhibitors (SNRIs), mirtazapine, reboxetine and agomelatine) and differences between individual SSRIs and TCAs. As antidepressants are largely similar in their therapeutic efficacy, awareness of profiles of unwanted effects is of particular importance.
Table 20.1 Classification of antidepressants
| Tricyclics | Selective serotonin reuptake inhibitors | Monoamine oxidase inhibitors |
|---|---|---|
| Dosulepin Amitriptyline Lofepramine Clomipramine Imipramine Trimipramine Doxepin Nortriptyline Protriptyline Desipramine | Fluoxetine Paroxetine Sertraline Citaloprama Escitaloprama Fluvoxamine | Phenelzine Isocarboxazid Tranylcypromine Moclobemide (RIMA) |
| Novel compounds | ||
|---|---|---|
| Mainly noradrenergic | Mainly serotonergic | |
| Reboxetine (NaRI) | Trazodoneb Nefazodoneb,c | |
| Mixed |
|---|
| Venlafaxine (SNRI) Mirtazapine (nassa)b Duloxetine (SNRI) Milnacipran (SNRI)d Agomelatine |
RIMA, reversible inhibitor of monoamine oxidase; NaRI, noradrenaline/norepinephrine reuptake inhibitor; SNRI, serotonin and noradrenaline/norepinephrine reuptake inhibitor; NaSSA, noradrenaline/norepinephrine and specific serotonergic antidepressant.
a Escitalopram is the active S-enantiomer of citalopram.
b Trazodone, nefazodone and mirtazapine have been classed as ‘receptor blocking’ antidepressants based on their antagonism of postsynaptic serotonin receptors (trazodone, nefazodone, mirtazapine) and presynaptic α2-receptors (trazodone, mirtazapine).
c Nefazodone has additional weak SSRI activity but has now been withdrawn due to risk of hepatitis.
An alternative categorisation of antidepressants is based solely on mechanism of action (Fig. 20.1). The majority of antidepressants, including SSRIs, TCAs and related compounds, are reuptake inhibitors. Certain novel agents, including trazodone and mirtazapine, are receptor blockers, whereas MAOIs are enzyme inhibitors.
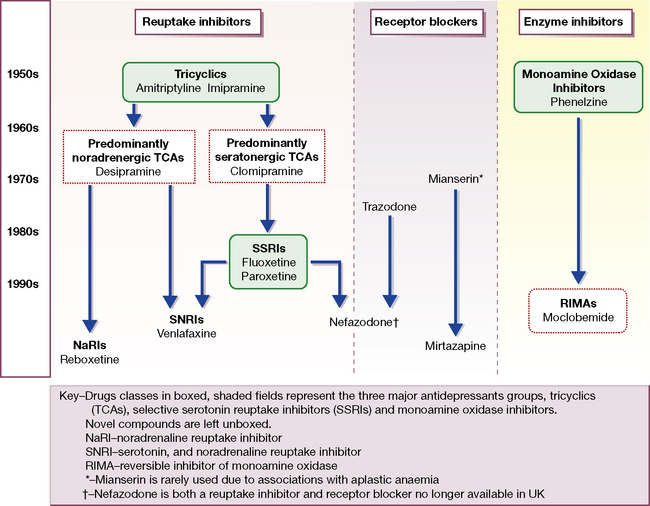
Fig. 20.1 Flow chart of the evolution of antidepressant drugs, and classification by mechanism of action.
The first TCAs (imipramine and amitriptyline) and MAOIs appeared between 1957 and 1961 (see Fig. 20.1). The MAOIs were developed from antituberculous agents that unexpectedly improved mood. Imipramine was a chlorpromazine derivative that showed antidepressant rather than antipsychotic properties. Over the next 25 years the TCA class enlarged to more than 10 agents with heterogeneous pharmacological profiles, and further modifications of the original three-ringed structure gave rise to the related (but pharmacologically distinct) antidepressant trazodone.
Mechanism of action
The monoamine hypothesis proposes that, in depression, there is deficiency of the neurotransmitters noradrenaline/norepinephrine and serotonin in the brain which can be restored by antidepressants. Drugs that alleviate depression also enhance monoamine availability and release (Fig. 20.2), increasing activity at postsynaptic receptors. It is relevant that (older) antihypertensive agents, e.g. reserpine, which reduced the availability of noradrenaline/norepinephrine, caused depression.
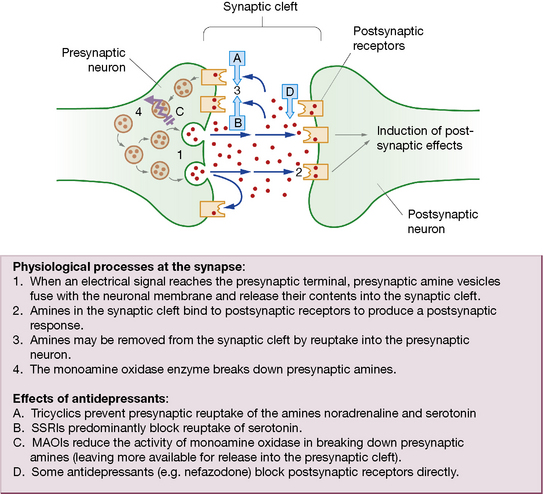
MAOIs increase the availability of noradrenaline/norepinephrine and serotonin by preventing their destruction by the monoamine oxidase type A enzyme in the presynaptic terminal (see Ch. 21, Table 21.3). The older MAOIs, phenelzine, tranylcypromine and isocarboxazid, bind irreversibly to monamine oxidase by forming strong (covalent) bonds. The enzyme is thus rendered permanently ineffective such that amine metabolising activity can be restored only by production of fresh enzyme, which takes weeks. These MAOIs are thus called ‘hit and run’ drugs as their effects greatly outlast their detectable presence in the blood.
Drugs with similar modes of action to antidepressants find other uses in medicine. Bupropion (amfebutamone) inhibits reuptake of both dopamine and noradrenaline/norepinephrine. It was originally developed and used as an antidepressant but is now more frequently used to assist smoking cessation (see p. 321). It is also prescribed in attention deficit hyperactivity disorder (see p. 345). Sibutramine, licensed as an anorectic agent, is a serotonin and noradrenaline/norepinephrine reuptake inhibitor (SNRI).
Pharmacokinetics
The antidepressants listed in Table 20.1 are generally well absorbed after oral administration. Steady-state plasma concentrations of TCAs show great individual variation but correlate with therapeutic effect. Where there is a failure of response, measurement of plasma concentration can be useful as the failure may be attributable to low plasma levels due to ultra-rapid metabolism (though it is often not available). Antidepressants in general are metabolised principally by hepatic cytochrome P450 enzymes. Of the many isoenzymes identified, the most important in antidepressant metabolism are CYP P450 2D6 (Table 20.2A) and CYP 3A4 (Table 20.2B). Other important P450 enzymes are CYP 1A2 (inhibited by the SSRI fluvoxamine, induced by cigarette smoking; substrates include caffeine and the atypical antipsychotics clozapine and olanzapine) and the CYP 2 C group (inhibition by fluvoxamine and fluoxetine, involved in breakdown of escitalopram and moclobemide). Sometimes several CYP enzymes are capable of mediating the same metabolic step. For example, at least six isoenzymes, including CYP 2D6, 3A4 and 2 C9, can mediate the desmethylation of the SSRI sertraline to its major metabolite.
Table 20.2A Psychotropic (and selected other) drugs known to be CYP 2D6 substrates, inhibitors and inducers
| CYP 2D6 inhibitors |
|---|
| Antidepressants |
| Paroxetine Fluoxetine |
| CYP 2D6 substrates | ||
|---|---|---|
| Antidepressants | Antipsychotics | Miscellaneous |
| Paroxetine Fluoxetine Citalopram Sertraline Venlafaxinea Duloxetine Amitriptyline Clomipramine Desipramine Imipramine Nortriptyline Reboxetine | Chlorpromazine Haloperidol Zuclopenthixol Perphenazine Risperidone | Dexfenfluramine Donepezil Opioids Codeine Hydrocodone Dihydrocodeine Tramadol Ethyl morphine MDMA (ecstasy) β-Blockers Propranolol Metoprolol Timolol Bufaralol Carvidelol |
A substrate is a substance that is acted upon and changed by an enzyme. Where two substrates of the same enzyme are prescribed together, they will compete and, if present in sufficient quantities, the metabolism of one or other, or both, drugs may also be inhibited, resulting in increased plasma concentration and possibly in enhanced therapeutic or adverse effects. An enzyme inducer accelerates the metabolism of co-prescribed drugs that are substrates of the same enzyme, reducing their effects. An enzyme inhibitor retards metabolism of co-prescribed drugs, increasing their effects.
a CYP 2D6 is involved only in the breakdown of venlafaxine to its active metabolite and therefore implications of 2D6 interactions for efficacy are of limited significance.
Table 20.2B Psychotropic (and selected other) drugs known to be CYP 3A4 substrates, inhibitors and inducers
| CYP 3A4 inhibitors | |
|---|---|
| Antidepressants | Other drugs |
| Fluoxetine Nefazodone | Cimetidine Erythromycin Ketoconazole (grapefruit juice) |
| CYP 3A4 substrates | ||
|---|---|---|
| Antidepressants | Anxiolytics, hypnotics and antipsychotics | Miscellaneous |
| Fluoxetine Sertraline Amitriptyline Imipramine Nortriptyline Trazodonea | Alpraxolam Aripiprazole Buspirone Diazepam Midazolam Triazolam Zoplicone Haloperidol Zuclopethixol Quetiapine Sertindole | Buprenorphine Carbamazepine Cortisol Dexamethasone Methadone Testosterone Calcium channel blockers Diltiazem Nifedipine Amlodipine Other drugs Amiodarone Omeprazole Oral contraceptives Simvastatin |
| CYP 3A4 inducers | |
|---|---|
| Antidepressants | Miscellaneous |
| St John’s wort | Carbamazepine Phenobarbital Phenytoin |
A substrate is a substance that is acted upon and changed by an enzyme. Where two substrates of the same enzyme are prescribed together, they will compete and, if present in sufficient quantities, the metabolism of one or other, or both, drugs may also be inhibited, resulting in increased plasma concentration and possibly in enhanced therapeutic or adverse effects. An enzyme inducer accelerates the metabolism of co-prescribed drugs that are substrates of the same enzyme, reducing their effects. An enzyme inhibitor retards metabolism of co-prescribed drugs, increasing their effects.
a mCPP (meta-chlorophenylpiperazine), the active metabolite of trazodone, is a CYP 2D6 substrate; observe for unwanted effects when trazodone is co-administered with the 2D6 inhibitors fluoxetine or paroxetine.
Therapeutic efficacy
• Venlafaxine, in high dose (> 150 mg/day) and Escitalopram may have greater efficacy than other antidepressants.
• Amitriptyline appears to be slightly more effective than other TCAs and also SSRIs for severe depression, but this advantage is compromised by its poor tolerability and lack of safety in overdose relative to more modern agents.
• The older MAOIs (e.g. phenelzine) may be more effective than other classes in ‘atypical’ depression, a form of depressive illness where mood reactivity is preserved, lack of energy may be extreme and biological features are the opposite of the normal syndrome, i.e. excess sleep and appetite with weight gain.
Changing and stopping antidepressants
When changing between antidepressant doses, a conservative approach would be to reduce the first antidepressant progressively over 2 or more weeks before starting the new drug. The gradual reduction is particularly important with paroxetine and venlafaxine which are known to cause ‘discontinuation syndromes’ if stopped abruptly, and less important with fluoxetine due to its long half-life active metabolite which offers ‘built-in’ protection against withdrawal problems. A more proactive approach would involve ‘cross-tapering’ the second antidepressant – i.e. starting it while the first antidepressant is being reduced and gradually titrating the dose up. However, an important exception concerns changes to or from MAOIs, which must be handled with great caution due to the dangers of interactions between antidepressants (see below). Therefore MAOIs cannot safely be introduced within 2 weeks of stopping most antidepressants (3 weeks for imipramine and clomipramine; combination of the latter with tranylcypromine is particularly dangerous), and not until 5 weeks after stopping fluoxetine, due to its long half-life active metabolite. Similarly, other antidepressants should not be introduced until 2–3 weeks have elapsed from discontinuation of MAOI (as these are irreversible inhibitors; see p. 313). No washout period is required when using the reversible MAOI, moclobemide.
Augmentation
Another important augmentation strategy employs the mood stabiliser lithium carbonate. Controlled trials suggest that up to 50% of patients who have not responded to standard antidepressants can respond after lithium augmentation but the evidence is stronger for augmenting tricyclics than for augmenting SSRIs. Addition of lithium requires careful titration of the plasma concentration up to the therapeutic range, with periodic checks thereafter and monitoring for toxicity (see p. 331).
More recently, augmentation of SSRIs with atypical antipsychotics has been effective in clinical trials. Trial evidence is strongest using olanzapine, and also exists for quetiapine, risperidone and aripiprazole. Antipsychotics also have important potential for side-effects which must be taken into account before their introduction (see p. 322).
Other indications for antidepressants
Antidepressants may benefit most forms of anxiety disorder, including panic disorder, generalised anxiety disorder, post-traumatic stress disorder, obsessive–compulsive disorder and social phobia (see p. 331). SSRIs are recognised as first-line drug treatment for all five of these anxiety disorders.
Adverse effects
Interactions
Pharmacodynamic interactions
• Most antidepressants (including SSRIs and tricylics) may cause central nervous system (CNS) toxicity if co-prescribed with the dopaminergic drugs entacapone and selegiline (for Parkinson’s disease). SSRIs increase the risk of the serotonin syndrome when combined with drugs that enhance serotonin transmission, e.g. the antimigraine triptan drugs which are 5HT1-receptor antagonists, and the antiobesity drug sibutramine.
• Most antidepressants lower the convulsion threshold, complicate the drug control of epilepsy and lengthen seizure time in electroconvulsive therapy (ECT). The situation is made more complex by the capacity of carbamazepine to induce the metabolism of antidepressants and of certain antidepressants to inhibit carbamazepine metabolism (see below).
• SSRIs are known to interfere with platelet aggregation and may increase the risk of gastrointestinal bleeding, especially in those with existing risk factors.
• Trazodone and many tricyclics cause sedation and therefore co-prescription with other sedative agents such as opioid analgesics, H1-receptor antihistamines, anxiolytics, hypnotics and alcohol may lead to excessive drowsiness and daytime somnolence.
• The majority of tricyclics have undesirable cardiovascular effects, in particular prolongation of the QTc interval. Numerous other drugs also prolong the QTc interval, e.g. amiodarone, disopyramide, procainamide, propafenone, quinidine, terfenadine, and psychotropic agents such as pimozide and sertindole. Their use in combination with TCAs that prolong QTc enhances the risk of ventricular arrhythmias.
• Tricylics potentiate the effects of catecholamines and other sympathomimetics, but not those of β2-receptor agonists used in asthma. Even the small amounts of adrenaline/epinephrine or noradrenaline/norepinephrine in dental local anaesthetics may produce a serious rise in blood pressure.
Pharmacokinetic interactions
Metabolism by cytochrome P450 enzymes provides ample opportunity for interaction of antidepressants with other drugs by inhibition of, competition for, or induction of enzymes. Tables 20.2A and 20.2B indicate examples of mechanisms by which interaction that may occur when relevant drugs are added to, altered in dose or discontinued from regimens that include antidepressants.
In depressive psychosis, antidepressants are commonly prescribed with antipsychotics and there is potential for enhanced drug effects with paroxetine + perphenazine (CYP 2D6), fluoxetine + sertindole (3A4) and fluvoxamine + olanzapine (1A2). Rapid tranquillisation with zuclopenthixol acetate (see p. 325) of an agitated patient who is also taking fluoxetine or paroxetine can result in toxic plasma concentrations with excessive sedation and respiratory depression due to inhibition of zuclopenthixol metabolism by CYP 2D6 and CYP 3A4. P450 enzyme inhibition by fluoxetine or paroxetine may also augment effects of alcohol, tramadol (danger of serotonin syndrome) methadone, terfenadine (danger of cardiac arrhythmia), -caine anaesthetics and theophylline.
e.g. carbamazepine and several other antiepilepsy drugs, accelerate the metabolism of antidepressants, reducing their therapeutic efficacy and requiring adjustment of dose. Epilepsy is a particularly common co-morbid illness in patients who have both psychiatric illness and learning disabilities, and the combination of an anticonvulsant and an antidepressant or major psychotropic drug is to be anticipated. Depression and hypertension are both such common conditions that some co-morbidity is inevitable. Panic disorder (an indication for an antidepressant drug) is associated with hypertension.1 An antidepressant that is an enzyme inhibitor may exaggerate antihypertensive effects of metoprolol (metabolised by CYP 2D6), or diltiazem or amlodipine (3A4).
Monoamine oxidase inhibitors
Foods likely to produce hypertensive effects in patients taking MAOIs include:
• Cheese, especially if well matured.
• Red wines (especially Chianti) and some white wines; some beers (non- or low-alcohol varieties contain variable but generally low amounts of tyramine).
• Yeast extracts (Marmite, Oxo, Bovril).
• Broad bean pods (contain dopa, a precursor of adrenaline/epinephrine).
• Over-ripe bananas, avocados, figs.
• Fermented bean curds including soy sauce.
• Fermented sausage, e.g. salami, shrimp paste.
MAOI interactions with other drugs
MAOI–SSRI combinations may provoke the life-threatening ‘serotonin syndrome’ (see above). Strict rules apply regarding washout periods when switching between MAOIs and other drugs (see above, Changing antidepressants, p. 316). Very occasionally, MAOIs are co-prescribed with other antidepressants, but as many combinations are highly dangerous such practice should be reserved for specialists only and then as a last resort.
St John’s wort
The herbal remedy St John’s wort (Hypericum perforatum) has found favour in some patients with mild to moderate depression. The active ingredients in the hypericum extract have yet to be identified, and their mode of action is unclear. Several of the known mechanisms of action of existing antidepressants are postulated, including inhibition of monoamine reuptake and the MAO enzyme, as well as a stimulation of GABA receptors. Much of the original research into the efficacy of St John’s wort was performed in Germany, where its use is well established. Several direct comparisons with tricyclic antidepressants have shown equivalent rates of response, but the interpretation of these studies is complicated by the fact that many failed to use standardised ratings for depressive symptoms, patients tended to receive TCAs below the minimum therapeutic dose, and sometimes received St John’s wort in doses above the maximum recommended in commercially available preparations. Use of St John’s wort is further complicated by the lack of standardisation of the ingredients. A large multi-centre trial found only limited evidence of benefit for St John’s wort over placebo in significant major depression.2
Antipsychotics
Classification
Originally tested as an antihistamine, chlorpromazine serendipitously emerged as an effective treatment for psychotic illness in the 1950s. Chlorpromazine-like drugs were originally termed ‘neuroleptics’ or ‘major tranquillisers’, but the preferred usage now is ‘antipsychotics’. Classification is by chemical structure, e.g. phenothiazines, butyrophenones. Within the large phenothiazine group, compounds are divided into three types on the basis of the side-chain, as this tends to predict adverse effect profiles (Table 20.3). The continuing search for greater efficacy and better tolerability led researchers and clinicians to reinvestigate clozapine, a drug that was originally licensed in the 1960s but subsequently withdrawn because of toxic haematological effects. Clozapine appeared to offer greater effectiveness in treatment-resistant schizophrenia, to have efficacy against ‘negative’ in addition to ‘positive’ psychiatric symptoms (see Table 20.4), and to be less likely to cause extrapyramidal motor symptoms. It regained its licence in the early 1990s with strict requirements on dose titration and haematological monitoring. The renewed interest in clozapine and its unusual efficacy and tolerability stimulated researchers to examine other ‘atypical’ antipsychotic drugs.
Table 20.3 Antipsychotic drugs
| Atypical antipsychoticsa | Classical antipsychotics | |
|---|---|---|
| Clozapine | Phenothiazines | |
| Olanzapine | Type 1 | Chlorpromazine |
| Quetiapine | Promazine | |
| Risperidone | Type 2 | Pericyazine |
| Ziprasidone | Type 3 | Trifluoperazine |
| Amisulprideb | Prochlorperazine | |
| Zotepine | Fluphenazine |



