Fig. 8.1
Information transfer in the central dogma of biology
Proteins perform vital functions in the body such as:
Catalyzing various biochemical reactions through enzymes.
Acting as messengers through neurotransmitters.
Acting as control elements which regulate cell reproduction.
Influencing growth and development of various tissues through trophic factors.
Transporting oxygen in the blood through hemoglobin.
Defending the body against diseases through antibodies.
The proteome changes constantly in response to tens of thousands of intra- and extracellular environmental signals. The proteome varies with health or disease, the nature of each tissue, the stage of cell development and effects of drug treatments. As such, the proteome often is defined as “the proteins present in one sample (tissue, organism, cell culture) at a certain point in time.”
The sequencing of the human genome has provided comprehensive resources for genomic data. In addition, the numerous fields such as transcriptomics, proteomics, and metabolomics, etc., and systems biology are providing new ways to study living processes. Genomics provides an overview of the complete set of genetic instructions provided by the DNA, while transcriptomics looks into gene expression patterns. Proteomics studies dynamic protein products and their interactions, while metabolomics is also an intermediate step in understanding organism’s entire metabolism (Fig. 8.2).
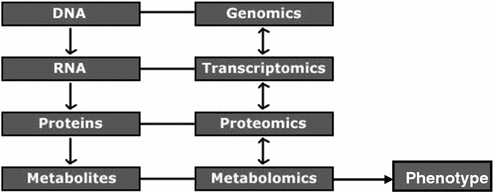
Fig. 8.2
Relationship between genomics, transcriptomics, proteomics, and metabolomics
Proteomics attempts to study the structure, function, and control of biological systems and processes by systematic and quantitative analysis of proteins. The term “proteomics” was first coined in 1997 to make an analogy with genomics, the study of the genes. The word “proteome” is a blend of “protein” and “genome”, and was coined by Marc Wilkins in 1994 while working on the concept as a Ph.D. student. Different definitions of proteomics have been given by different workers from time to time as, the study of full set of proteins encoded by the genome or the study of all the proteins expressed in a cell.
As the genome describes the genetic content of an organism, a proteome defines the protein complement of the genome. The proteome is dynamic, and is the set of proteins expressed in a specific cell, given a particular set of conditions. Proteomics begins with the functionally modified protein and works back to the gene responsible for its production.
8.1.1 Structure of Proteins
Proteins can be organized in four structural levels:
1.
Primary—The amino acid sequence, containing members of 20 amino acids.
2.
Secondary—Local folding of the amino acid sequence into α helices and β sheets.
3.
Tertiary—3D conformation of the entire amino acid sequence.
4.
Quaternary—Interaction between multiple small peptides or protein subunits to create a large unit.
Each level of protein structure is essential to the biological function of the protein. The primary sequence of the amino acid chain determines where secondary structures will form, as well as the overall shape of the final 3D conformation. The 3D conformation of each small peptide or subunit determines the final structure and function of the protein (Garret and Grisham 1995).
The proteome is the entire complement of proteins. It is now known that mRNA is not always translated into protein, and the amount of protein produced for a given amount of mRNA depends on the gene it is transcribed from and on the current physiological state of the cell. Proteomics confirms the presence of the protein and provides a direct measure of the quantity present. The level of transcription of a gene gives only a rough estimate of its level of expression into a protein. A mRNA produced in abundance may be degraded rapidly or translated inefficiently, resulting in a small amount of protein. Many proteins experience post-translational modifications that profoundly affect their activities and many transcripts give rise to more than one protein, through alternative splicing or alternative post-translational modifications. Further, many proteins form complexes with other proteins or RNA molecules, and only function in the presence of these other molecules and finally, protein degradation rate plays an important role in protein content.
8.1.2 Broad-Based Proteomics
The first step when utilizing broad-based proteomics is to develop a hypothesis specific to the proteome being studied. For this an organism that already has a great deal of genomic information available is taken, since the genome is always a useful supplement to proteomic information. Then the technologies are chosen which should be compatible with the sample. Some proteomic methods include High-performance liquid chromatography (HPLC), Mass Spectrometry, SDS-PAGE, two-dimensional gel electrophoresis, and in silico protein modeling. There are many sample type, sample preparation, and analytical technology combinations which can be used for study in proteomics. Table 8.1 compares the broad-based approach with traditional focused approach in proteomics.
Table 8.1
Broad-based proteomics approach versus traditional focused approach
Broad-based approach | Focussed approach | |
|---|---|---|
Goal | Understand the proteome as a whole | Understand specific protein function |
Basic steps | 1. Identify organism | 1. Identify protein |
2. Understand sample type and preparation | 2. Understand sample type and preparation | |
3. Utilize analytical technology compatible with sample type | 3. Isolate protein | |
4. Bioinformatic analysis of the proteome sample | 4. Utilize analytical technology that is compatible | |
5. Build a proteomic model | 1. Bioinformatic analysis of the protein sample | |
2. Model protein’s function and/or structure | ||
Pros | 1. Proteomic information about a specific tissue under certain conditions can be gained | 1. Inexpensive and results can be generated much quicker than a large proteomic study |
2. Relationships between many proteins can be understood | 1. Functional and structural information about a protein can be determined | |
Cons | 1. Extensive upfront planning | 1. Protein information may not be valuable without a global proteomic understanding |
2. The study will cost more and last longer than a focused study | 2. Hard to develop global relationships from many focused experiments taken together | |
3. No guarantee that quality proteomic data will be generated in the end | ||
Common technologies implemented | 1. SDS-PAGE | 1. SDS-PAGE |
2. 2DE-DIGE | 2. HPLC | |
3. HPLC | 3. Mass spectroscopy (MS) | |
4. Mass spectroscopy (MS) | 4. Molecular modeling tools (bioinformatics) | |
5. Proteomic modeling tools (bioinformatics) |
8.1.3 Human Proteome Organization
Human Proteome Organization (HUPO) is an international scientific organization representing and promoting proteomics through international cooperation and collaborations by fostering the development of new technologies, techniques, and training. It is an international consortium of national proteomics research associations, government researchers, academic institutions, and industry partners. HUPO, founded in June 2001, promotes the development and awareness of proteomics research, advocates proteomics researchers throughout the world, and facilitates scientific collaborations between HUPO members and initiatives, organized to gain better and complete understanding of the human proteome. The Human Proteome Organization is currently working on establishing a defined standard for data submission and annotation for the many different proteomics techniques currently used to identify and annotate proteins. The proteomics standards initiative (PSI) is a working group of HUPO. It aims to define data standards for proteomics in order to facilitate data comparison, exchange, and verification. PSI focuses on the following:
Minimum Information about a Proteomics Experiment (MIAPE) defines the metadata that should be provided along with a proteomics experiment.
Data Markup Languages for encoding the data and metadata.
Ontologies for consistent annotation and representation.
8.2 Classification of Proteomics
Proteomics is mainly concerned with determining the structure, expression, localization, biochemical activity, interactions, and cellular roles of proteins. According to Graves and Haystead (2002) proteomics can be broadly classified into three types:
1.
Structural proteomics—It is in-depth large-scale analysis of protein structure. It deals with determination of the 3D-structure of protein complexes or the proteins present in a specific cellular organelle. Protein structure comparisons can help to identify the functions of newly discovered genes. Structural analysis can also show where drugs bind to proteins and where proteins interact with each other. This is achieved using technologies such as X-ray crystallography and NMR spectroscopy.
2.
Expression proteomics—Large-scale analysis of expression and differential expression of proteins is called as expression proteomics. This can help to identify the main proteins found in a particular sample and proteins differentially expressed in related samples, such as diseased versus healthy tissue. A protein found only in a diseased sample may represent a useful drug target or diagnostic marker. Proteins with similar expression profiles may also be functionally related. Technologies such as 2D-PAGE and mass spectrometry are used for this.
3.
Interaction proteomics—It deals with analysis of interactions between proteins to characterize complexes and determine function. The characterization of protein–protein interactions helps to determine protein functions and can also show how proteins assemble in larger complexes. Technologies such as affinity purification, mass spectrometry, and the yeast two-hybrid system are particularly useful for this.
8.3 Basic Steps in Proteomics
The following steps are involved in a proteomics experiment (Fig. 8.3):
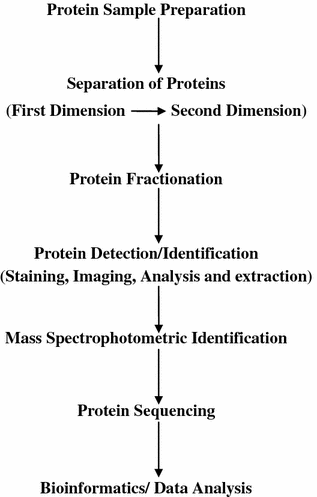
Fig. 8.3
Flow diagram of the steps involved in proteomics
1.
protein isolation from a biological sample (e.g., a cell extract) following some experimental treatment.
2.
fractionation of the resulting proteins (or peptides, the products of proteome digestion) by methods such as two-dimensional polyacrylamide gel electrophoresis (2D-PAGE) or liquid chromatography (LC).
3.
protein or peptide detection by MS.
4.
protein identification through manual interpretation or database correlation of mass spectra.
8.3.1 Protein Sample Preparation
With technological advances in proteomics, procedures for preparation of protein samples prior to any particular procedure have also advanced. A number of issues arise in this respect, including sample clean-up, fractionation, enrichment, and also sample condition optimization. This facet of proteomics is becoming particularly critical in case of high-throughput protocols where the necessary conditions of a sample in one stage may directly conflict with the efficacy of a second stage. For example, during the initial step in 2D electrophoresis and isoelectric focusing, all proteins in a sample are given a net charge of zero; while in the second step, gel electrophoresis requires a negative charge on all products in the sample in order to induce movement through the gel matrix. Many companies, e.g., Millipore, BioRad, Gelifesciences, Invitrogen, Aligent, Beckmancoulter, Bioproximity, etc., offer pre-packaged kits that allow to prepare samples for many different techniques. They also offer many protein samples and other protein technologies.
8.3.2 Determining the Existence of Proteins in Complex Mixtures
Classically, antibodies to particular proteins or to their modified forms have been used in biochemistry and cell biology studies. For more quantitative determinations of protein amounts, techniques such as ELISAs can be used. For proteomic study, more recent techniques such as Matrix-assisted laser desorption or ionization has been employed for rapid determination of proteins in particular mixtures.
8.3.3 Determining Post-translationally Modified Proteins
A particular protein can be studied by developing an antibody which is specific to that modification. For example, there are antibodies which only recognize certain proteins when they are tyrosine-phosphorylated, also, there are antibodies specific to other modifications. These can then be used to determine the set of proteins that have undergone the modification of interest. For sugar modifications, such as glycosylation of proteins, certain lectins have been discovered which bind sugars. A more common way to determine post-translational modification of interest is by 2D gel electrophoresis. Recently, another approach called PROTOMAP has been developed which combines SDS-PAGE with shotgun proteomics to enable detection of changes in gel-migration such as those caused by proteolysis or post-translational modification.
8.4 Techniques Used in Proteomics
Some of the techniques used in proteomics study are given below:
1.
One- and two-dimensional gel electrophoresis is used to identify the relative mass of a protein and its isoelectric point.
2.
HPLC is used for separation of proteins.
3.
X-ray crystallography and nuclear magnetic resonance are used to characterize the 3D structure of peptides and proteins. However, low-resolution techniques such as circular dichroism, Fourier transform infrared spectroscopy, and small angle X-ray scattering can be used to study the secondary structure of proteins.
4.
Tandem mass spectrometry (MS/MS) combined with reverse phase chromatography or 2D electrophoresis is used to identify (by de novo peptide sequencing) and quantify all the levels of proteins found in cells.
5.
Mass spectrometry, often MALDI-TOF, is used to identify proteins by peptide mass fingerprinting (PMF). Less commonly this approach is used with chromatography and/or high-resolution mass spectrometry.
6.
Affinity chromatography, Yeast two-hybrid techniques, Fluorescence resonance energy transfer (FRET), and Surface plasmon resonance (SPR) are used to identify protein–protein and protein-DNA binding reactions.
7.
X-ray tomography is used to determine the location of labeled proteins or protein complexes in an intact cell. It is frequently correlated with images of cells from light-based microscopes.
8.
Software-based image analysis is utilized to automate the quantification and detection of spots within and among gel samples.
Some of these techniques have been discussed in detail.
8.4.1 One- and Two-Dimensional Gel Electrophoresis
One- and two-dimensional gel electrophoresis is used to identify the relative mass and isoelectric point of a protein.
8.4.1.1 Electrophoresis
The migration of charged colloidal particles or molecules through a solution under the influence of an applied electric field is usually provided by immersed electrodes. It can also be described as a method of separating substances, especially proteins and analyzing molecular structure based on the rate of movement of each component in a colloidal suspension under the influence of an electric field. An analyte is a chemical substance that is the subject of chemical analysis. Separation by electrophoresis depends on differences in the migration velocity of ions or solutes through a given medium in an electric field. The electrophoretic migration velocity of an analyte is 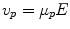 where E is the electric field strength and
where E is the electric field strength and 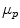 is the electrophoretic mobility.
is the electrophoretic mobility.
where E is the electric field strength and is the electrophoretic mobility.The electrophoretic mobility is inversely proportional to frictional forces in the buffer, and directly proportional to the ionic charge of the analyte. The forces of friction against an analyte are dependent on the analyte’s size and the viscosity (η) of the medium. Analytes with different frictional forces or different charges will separate from one another when they move through a buffer. At a given pH, the electrophoretic mobility of an analyte is:
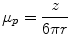 where, r is the radius of the analyte and z is the net charge of the analyte.
where, r is the radius of the analyte and z is the net charge of the analyte.
Differences in the charge to size ratio of analyte cause differences in electrophoretic mobility. Small, highly charged analytes have greater mobility, whereas large, less charged analytes have lower mobility. Electrophoretic mobility is an indication of an analyte’s migration velocity in a given medium. The net force acting on an analyte is the balance of two forces: the electrical force acting in favor of motion, and the frictional force acting against motion. The two forces remain steady during electrophoresis, thus the electrophoretic mobility is a constant for a given analyte under a given set of conditions.
Electrophoresis has a wide variety of applications in proteomics, forensics, molecular biology, genetics, biochemistry, and microbiology. One of the most common uses of electrophoresis is to analyze differential expression of genes. Healthy and diseased cells can be identified by differences in the electrophoretic patterns of their proteins. Proteins can also be characterized similarly, and some information about their structure can be derived from the masses of fragments in the gel.
8.4.1.2 Two-Dimentional Gel Electrophoresis
Two-dimensional polyacrylamide gel electrophoresis (2DE) was first described by O’Farrell in 1975 and has evolved markedly as one of the core technologies for the analysis of complex protein mixtures extracted from biological samples since then. The proteins are separated in 2 steps according to 2 independent properties [isoelectric point (pI) and molecular weight (MW)].
The proteins are made up of amino acids which may have positive, negative, or no charges. In addition, amino acids can be hydrophobic or hydrophilic. These amino acid properties are combined in the molecule to determine the electrostatic and amphiphilic properties of the proteins. When a protein of charge q is placed in an electric field, E, it experiences an electrical force given by: F = q × E. Under the influence of this force, the protein moves until the force becomes zero (F = 0). This principle is used to make proteins move in a liquid or solid media. A common medium used is Polyacrylamide (PAA). PAA is a flexible, elastic polymer that allows particles to move inversely proportional to their sizes; i.e., smaller particles move faster than larger particles. Thus a mixture of proteins in a PAA gel under the influence of a force F will separate in individual molecules depending on their sizes and charges. Two-dimensional polyacrylamide gel electrophoresis (2D-PAGE) is a form of gel electrophoresis in which proteins are separated and identified in two dimensions oriented at right angles to each other. Small changes in charge and mass can easily be detected by this method, because it is rare that two different proteins will resolve to the same place in both dimensions. A 2D gel can resolve one thousand to two thousand proteins, which appear, after staining, as dots in the gel.
The three main advantages of this technique are its robustness, its parallelism, and its unique ability to analyze complete proteins at high resolution. This technique is useful when comparing two similar samples to find specific protein differences. Using 2D-PAGE, hundreds to thousands of polypeptides can be analyzed in a single run. The proteins can be separated in pure form from the resultant spots which can be quantified and further analyzed by mass spectrometry, depending on their resolution. Polypeptides can also be probed with antibodies and tested for post-translational modifications. 2D-PAGE is also used to study differential expression of proteins between cell types. The two main drawbacks are its very low efficiency in the analysis of hydrophobic proteins, and high sensitivity toward the dynamic range and quantitative distribution issues. It requires a large amount of sample handling, limited reproducibility, and a smaller dynamic range than other separation methods. It is also not automated for high-throughput analysis. Certain proteins are difficult for 2D-PAGE to separate such as those that are in low abundance, acidic, basic, hydrophobic, very large, or very small.
8.4.2 Centrifugation
Centrifugation is one of the most important and widely applied research techniques in biochemistry, cellular and molecular biology, and in medicine. In proteomics it plays a vital role in the fundamental and necessary process of isolating proteins. This process begins with intact cells or tissues. Before the proteins can be obtained, the cells must be broken open by processes such as snap freezing, sonication, homogenization by high pressure, or grinding with liquid nitrogen. Once the cells have been opened up, all of their contents including cell membranes, RNA, DNA, and organelles will be mixed in the solvent with the proteins. Centrifugation is used for separating out all the non-protein material. Within the centrifuge samples are spun at high speeds and the resulting force causes particles to separate based on their density. Centrifugation is also used for removing cells or other suspended particles from their surroundings, isolating viruses and macromolecules, including DNA, RNA, proteins, and lipids or establishing physical parameters of these particles from their observed behavior during centrifugation, separating from dispersed tissue the various subcellular organelles including nuclei, mitochondria, chloroplasts, golgi bodies, lysosomes, peroxisomes, glyoxysomes, plasma membranes, endoplasmic reticulum, polysomes, and ribosomal subunits. Once the mixture of proteins has been isolated using centrifugation, one of several methods to separate out individual proteins can be used for further study.
8.4.3 Protein Separation-Chromatography
To obtain a pure protein sample, a protein has to be isolated from all other proteins and cellular components. This can prove to be difficult because a single protein often makes up only 1 % of the total protein concentration of a cell. Therefore, 99 % of the protein components of a sample must be removed before it can be classified as pure. Protein separations can be done by chromatography. There are several properties of proteins that can be used to separate them. Different types of chromatography take advantage of different properties. Proteins can be separated on the basis of size, shape, hydrophobicity, affinity to molecules or charge. All methods utilize an insoluble stationary phase and a mobile phase that passes over it. The mobile phase is commonly a liquid solution which contains the protein that has to be isolated. The stationary phase on the other hand is made up of a group of beads, usually based on a carbohydrate or acrylamide derivative, that are bound to ionically charged species, hydrophobic characters, or affinity ligands.
In column chromatography, when a protein sample is applied to the column, it equilibrates between the stationary phase and the mobile phase. Depending on the type of chromatography, proteins with certain characteristics will bind to the stationary phase while those lacking the sought characteristics will remain in the mobile phase and pass through the column. For example in ion exchange chromatography, a positively charged protein binds to a negatively charged stationary phase, while the negatively charge protein will be eluted from the column with the mobile phase. The final step involves displacing the protein from the stationary phase, also known as elution. This is done by introducing a particle which will compete with the protein binding site on the stationary phase. Various commercial columns are available; specifically Bio-Rad, Sigma-Aldrich, GE Healthcare, etc., offer a variety of chromatography columns (Fig. 8.4).
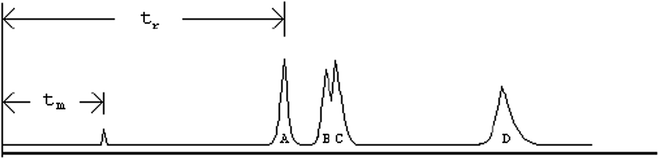
Fig. 8.4
Chromatogram showing separation based on signals given by a detector. X axis: Time in Min or volume in ml. Y axis: Signal. t m — the time required for the mobile phase to travel the entire length of the column, t r —the time required for a specific protein to elute from the column
8.4.4 Emerging and Miscellaneous Technologies in Proteomics
8.4.4.1 Multi-dimentional Protein Identification Technology or Orthogonal Separations
Multi-dimentional protein identification technology (MudPIT) is a chromatography-based proteomic technique in which a complex peptide mixture is prepared from a protein sample and loaded directly onto a triphasic microcapillary column packed with reversed phase, strong cation exchange, and reversed phase HPLC grade materials. Once the complex peptide mixture is loaded the column is placed directly in-line with a tandem mass spectrometry (MS/MS). The MS/MS data generated from a MudPIT run is then searched to determine the protein content of the original sample.
MudPIT is a robust and widely accepted method for protein identification from a wide variety of samples. It is an excellent tool for both qualitative and quantitative proteomic analyses. Through on-line 2D HPLC, complex peptides mixtures can be well separated. For a relatively simple sample, better results are obtained from MudPIT, compared to single dimension liquid chromatography method. It was developed as a method to analyze the highly complex samples necessary for large-scale proteome analysis by electrospray ionization, MS/MS, and database searching. This method couples a 2D liquid chromatography separation of peptides on a microcapillary column with detection in a tandem mass spectrometer. In the MudPIT, a protein or mixture of proteins is first reduced (to break cysteine disulfide bonds), alkylated (to prevent reformation of disulfide bonds), and digested into a complex mixture of peptides.
MudPIT has been used in a wide range of proteomics experiments, including large-scale catalogs of proteins in cells and organisms, profiling of organelle and membrane proteins, identification of protein complexes, determination of post-translational modifications and quantitative analysis of protein expression. In MudPIT biochemical fractions containing many proteins are directly proteolyzed and the enormous number of peptides generated, are separated by 2D liquid chromatography before entering the mass spectrometer. Instead of MALDI-TOF, MS/MS is employed so that, after the mass of a peptide is measured, the peptide is fragmented using a collision-induced dissociation cell, and the masses of the fragmentation products are determined.
8.4.4.2 Isotope-Coded Affinity Tag
Isotope-coded affinity tags (ICATs) are gel-free method for quantitative proteomics that relies on chemical labeling reagents. These chemical probes consist of three general elements: a reactive group capable of labeling a defined amino acid side chain (e.g., iodoacetamide to modify cysteine residues), an isotopically coded linker and a tag (e.g., biotin) for the affinity isolation of labeled proteins/peptides. For the quantitative comparison of two proteomes, one sample is labeled with the isotopically light (d0) probe and the other with the isotopically heavy (d8) version (Fig. 8.5). To minimize error, both samples are then combined, digested with a protease (i.e., trypsin), and subjected to avidin affinity chromatography to isolate peptides labeled with isotope-coded tagging reagents. These peptides are then analyzed by liquid chromatography–mass spectrometry (LC–MS). The ratios of signal intensities of differentially mass-tagged peptide pairs are quantified to determine the relative levels of proteins in the two samples. The original tags were developed using deuterium, but later tags using 13C were used instead to circumvent issues of peak separation during liquid chromatography due to the interaction of deuterium with the stationary phase of the column.
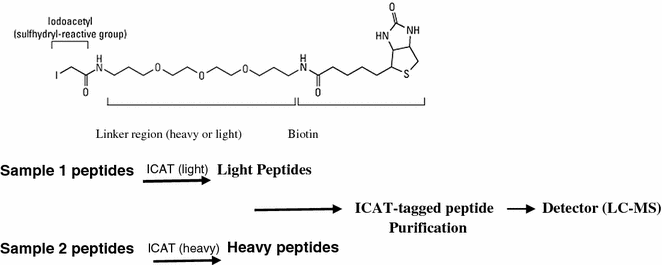
Fig. 8.5
Process of ICAT used in proteomics
For samples that are not amenable to metabolic labeling, such as when analyzing clinical samples (e.g., biological fluids, tissue samples) or when experimental time is limited, chemical or enzymatic stable isotopic labeling methods are available for quantitative proteomic analyses. These include strategies to add isotopic atoms or isotope-coded tags to peptides or proteins. A rapid and relatively inexpensive method of chemical labeling is stable isotope dimethylation which uses formaldehyde in deuterated water to label primary amines with deuterated methyl groups. This approach also does not change the ionic state of the labeled peptides because of the reductive amination that occurs, so their chemical properties remain the same as those of unlabeled peptides. Benefit of this approach is that many samples are amenable to formaldehyde fixation, which is fast and cheap compared to other labeling reagents. This requires using pure samples or sample preparation to reduce the complexity of biological samples to minimize the number of peaks detected by MS.
Protein labeling with ICAT followed by MS/MS allows sequence identification and accurate quantification of proteins in complex mixtures, and has been applied to the analysis of global protein expression changes, protein changes in subcellular fractions, components of protein complexes, protein secretion, and body fluids.
8.4.4.3 Label-Free Tags
Label-free methods for both relative and absolute quantitation have been developed as a rapid and low-cost alternative to other quantitative proteomic approaches. These strategies are ideal for large-sample analyses in clinical screening or biomarker discovery experiments but are less reliable for measuring small changes. Unlike other quantitation methods, label-free samples are separately collected, prepared, and analyzed by liquid chromatography–mass spectrometry, LC–MS, or LC–MS/MS. Hence, label-free quantitation experiments need to be more carefully controlled than stable isotope methods to account for any experimental variations. Protein quantitation is performed using either ion peak intensity or spectral counting.
Relative quantitation by ion peak intensity relies on LC–MS only. The direct MS m/z values for all ions are detected and their signal intensities at a particular time recorded. The signal intensity from electrospray ionization has been reported to highly correlate with ion concentration, therefore the relative peptide levels between samples can be determined directly from these peak intensities. Because of the large amount of data collected from these experiments, sensitive computer algorithms are required for automated ion peak alignment and comparison. Label-free relative quantitation by spectral counts entails comparing the sum of the MS/MS spectra from a given peptide across multiple samples, which has been shown to directly correlate with protein abundance. Besides relative quantitation, label-free methods can be used to determine the absolute concentration of proteins in a sample. One method entails determining the exponentially modified protein abundance index (emPAI), which estimates protein abundance based on the number of peptides detected and the number of theoretically observed tryptic peptides for each protein, is used to determine the approximate absolute protein abundance in large-scale proteomic analyses. Another method, absolute protein expression (APEX) is based on spectral counts and uses correction factors to make protein abundance proportional to the number of peptides observed.
8.4.4.4 Flourescence Resonance Energy Transfer
Flourescence resonance energy transfer (FRET) is an important tool to study protein–protein interactions, protein-DNA interactions, protein conformational changes and other molecular dynamics quantification. It is used to detect protein binding, providing spatial and temporal information about the interaction. It is a type of Fluorescence Spectroscopy using two fluorescent dyes with overlapping emission and absorption spectra, which is used to indicate proximity of labeled molecules. This technique is useful for studying interactions of molecules and protein folding. It uses fluorescent energy transfer to visualize protein interactions. Fluorophores are fused to the proteins of interest and then bombarded with light at the excitation wavelength. The first fluorophore transfers some of the energy it absorbs from the light source to the second fluorophore, which in turn emits some of its energy into the environment where it is visible with the use of a fluorescent microscope.
Since the discovery of FRET by Theodor Forster in 1948, it has become useful tool in biology for three reasons: (1) FRET is really sensitive in the range of 100 Å and below, the scale at which transactions between biological macromolecules and complexes occur; (2) the instrumentation is extremely sensitive and is readily amenable to miniaturization, high throughput, and automation; and (3) cell permeable and genetically encodable FRET probes enable the real‐time quantitation of dynamic cellular processes in live cells.
8.4.4.5 Mass Spectrometry
Mass spectrometry (MS) is an important emerging method for the characterization of proteins. MS is a technique in which gas phase molecules are ionized and their mass-to-charge ratio is measured by observing acceleration differences of ions when an electric field is applied (Fig. 8.6). Lighter ions will accelerate faster and be detected first. If the mass is measured with precision then the composition of the molecule can be identified. In the case of proteins, the sequence can be identified. Most samples submitted to MS are a mixture of compounds. A spectrum is acquired to give the mass-to-charge ratio of all compounds in the sample. MS throws light on molecular mechanisms within cellular systems. It is used for identifying proteins, functional interactions, and it further allows for determination of subunits. Several configurations of mass spectrometers that combine electrospray ionization (ESI) and matrix-assisted laser desorption ionization (MALDI) with a variety of mass analyzers (linear quadrupole mass filter [Q], time-of-flight [ToF], quadrupole ion trap, and Fourier transform ion cyclotron resonance [FTICR] instrument) are routinely used (Yates et al. 2009).
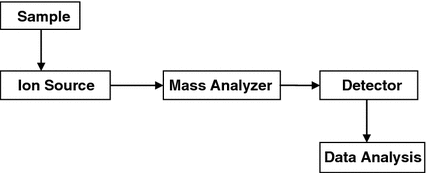
Fig. 8.6
Schematic diagram of a mass spectrometer
Whole protein mass analysis is primarily conducted using either TOF, MS, or FTICR. These two instruments are preferable because of their wide mass range and in the case of FTICR, its high-mass accuracy. Mass analysis of proteolytic peptides is a much more popular method of protein characterization, as cheaper instrument designs can be used for characterization. Additionally, sample preparation is easier once whole proteins have been digested into smaller peptide fragments. The most widely used instrument for peptide mass analysis are the MALDI time-of-flight instruments as they permit the acquisition of peptide mass fingerprints (PMFs) at high pace. Multiple stage quadrupole-time-of-flight and the quadrupole ion trap also find use in this application.
The relative abundance of an ion can also be measured using MS. Different compounds have differential ionization capabilities, therefore intensity of an ion is not in a direct correlation to concentration. It is an analytical method which has a variety of uses outside of proteomics, such as isotope and dating, trace gas analysis, atomic location mapping, pollutant detection, and space exploration. This technique was discovered during studies of gas excitation in a charged environment, more than 100 years ago by J. J. Thomson in 1913.
The ionization methods used for the majority of biochemical analyses are:
1.
ESI
ESI is one of the atmospheric pressure ionization (API) techniques and is well suited to the analysis of polar molecules ranging from less than 100 Da to more than 1,000,000 Da in molecular mass. In ESI the ions of interest are formed from solution by applying a high electric field to the tip of a capillary, from which the solution will pass through. The sample will be sprayed into the electric field along with a flow of nitrogen to promote desolvation. Droplets will form and will evaporate in a vacuumed area. This causes an increase in charge on the droplets and the ions are now said to be multiply charged. These multiply charged ions can then enter the analyzer (Andersen et al. 1996). ESI is a method of choice because of the following properties: (1) The “softness” of the phase conversion process allows very fragile molecules to be ionized intact and even in some non-covalent interactions to be preserved for MS analysis. (2) The eluting fractions through liquid chromatography can then be sprayed into the mass spectrometer, allowing for the further analysis of mixtures. (3) The production of multiply charged ions allow for the measurement of high-mass biopolymers. Multiple charges on the molecule will reduce its mass to charge ratio when compared to a single charged molecule. Multiple charges on a molecule also allow for improved fragmentation which in turn allows for a better determination of structure.
2.
MALDI
MALDI (Hillenkamp et al. 1991) deals with thermolabile, non-volatile organic compounds especially those of high molecular mass and is used successfully in biochemical areas for the analysis of proteins, peptides, glycoproteins, oligosaccharides, and oligonucleotides. It is relatively straightforward to use and reasonably tolerant to buffers and other additives. The mass accuracy depends on the type and performance of the analyzer of the mass spectrometer, but most modern instruments are capable of measuring masses to within 0.01 % of the molecular mass of the sample, at least up to ca. 40,000 Da. In MALDI, the molecular ions of interest are formed by pulses of laser light impacting on the sample isolated within an excess of matrix molecules. This enables the determination of masses of large biomolecules and synthetic polymers greater than 200,000 Daltons without degradation of the molecule of interest. The advantages of MALDI are its robustness, high speed, and relative immunity to contaminants and biochemical buffers. A type of mass spectrometer often used with MALDI is TOF or Time-of-Flight mass spectrometry. This enables fast and accurate molar mass determination along with sequencing repeated units and recognizing polymer additives and impurities. This technique is based on an ultraviolet absorbing matrix where the matrix and polymer are mixed together along with excess matrix and a solvent to prevent aggregation of the polymer. This mixture is then placed on the tip of a probe; then the solvent is removed while under vacuum conditions. This creates co-crystallized polymer molecules that are dispersed homogeneously within the matrix. A pulsing laser beam is set to an appropriate frequency and energy is shot to the matrix, which becomes partially vaporized. As a result the homogeneously dispersed polymer within the matrix is carried into the vapor phase and becomes charged.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


