5
Proteins: Higher Orders of Structure
Peter J. Kennelly, PhD & Victor W. Rodwell, PhD
OBJECTIVES
After studying this chapter, you should be able to:
![]() Indicate the advantages and drawbacks of several approaches to classifying proteins.
Indicate the advantages and drawbacks of several approaches to classifying proteins.
![]() Explain and illustrate the primary, secondary, tertiary, and quaternary structure of proteins.
Explain and illustrate the primary, secondary, tertiary, and quaternary structure of proteins.
![]() Identify the major recognized types of secondary structure and explain supersecondary motifs.
Identify the major recognized types of secondary structure and explain supersecondary motifs.
![]() Describe the kind and relative strengths of the forces that stabilize each order of protein structure.
Describe the kind and relative strengths of the forces that stabilize each order of protein structure.
![]() Describe the information summarized by a Ramachandran plot.
Describe the information summarized by a Ramachandran plot.
![]() Indicate the present state of knowledge concerning the stepwise process by which proteins are thought to attain their native conformation.
Indicate the present state of knowledge concerning the stepwise process by which proteins are thought to attain their native conformation.
![]() Identify the physiologic roles in protein maturation of chaperones, protein disulfide isomerase, and peptidylproline cis-trans-isomerase.
Identify the physiologic roles in protein maturation of chaperones, protein disulfide isomerase, and peptidylproline cis-trans-isomerase.
![]() Describe the principal biophysical techniques used to study tertiary and quaternary structure of proteins.
Describe the principal biophysical techniques used to study tertiary and quaternary structure of proteins.
![]() Explain how genetic and nutritional disorders of collagen maturation illustrate the close linkage between protein structure and function.
Explain how genetic and nutritional disorders of collagen maturation illustrate the close linkage between protein structure and function.
![]() For the prion diseases, outline the overall events in their molecular pathology and name the life forms each affects.
For the prion diseases, outline the overall events in their molecular pathology and name the life forms each affects.
BIOMEDICAL IMPORTANCE
In nature, form follows function. In order for a newly synthesized polypeptide to mature into a biologically functional protein capable of catalyzing a metabolic reaction, powering cellular motion, or forming the macromolecular rods and cables that provide structural integrity to hair, bones, tendons, and teeth, it must fold into a specific three-dimensional arrangement, or conformation. In addition, during maturation post-translational modifications may add new chemical groups or remove transiently-needed peptide segments. Genetic or nutritional deficiencies that impede protein maturation are deleterious to health. Examples of the former include Creutzfeldt–Jakob disease, scrapie, Alzheimer’s disease, and bovine spongiform encephalopathy (“mad cow disease”). Scurvy represents a nutritional deficiency that impairs protein maturation.
CONFORMATION VERSUS CONFIGURATION
The terms configuration and conformation are often confused. Configuration refers to the geometric relationship between a given set of atoms, for example, those that distinguish L– from D-amino acids. Interconversion of configurational alternatives requires breaking (and reforming) covalent bonds. Conformation refers to the spatial relationship of every atom in a molecule. Interconversion between conformers occurs without covalent bond rupture, with retention of configuration, and typically via rotation about single bonds.
PROTEINS WERE INITIALLY CLASSIFIED BY THEIR GROSS CHARACTERISTICS
Scientists initially approached structure–function relationships in proteins by separating them into classes based upon properties such as solubility, shape, or the presence of nonprotein groups. For example, the proteins that can be extracted from cells using aqueous solutions of physiologic pH and ionic strength are classified as soluble. Extraction of integral membrane proteins requires dissolution of the membrane with detergents. Globular proteins are compact, roughly spherical molecules that have axial ratios (the ratio of their shortest to longest dimensions) of not over 3. Most enzymes are globular proteins. By contrast, many structural proteins adopt highly extended conformations. These fibrous proteins possess axial ratios of 10 or more.
Lipoproteins and glycoproteins contain covalently bound lipid and carbohydrate, respectively. Myoglobin, hemoglobin, cytochromes, and many other metalloproteins contain tightly associated metal ions. While more precise classification schemes have emerged based upon similarity, or homology, in amino acid sequence and three-dimensional structure, many early classification terms remain in use.
PROTEINS ARE CONSTRUCTED USING MODULAR PRINCIPLES
Proteins perform complex physical and catalytic functions by positioning specific chemical groups in a precise three-dimensional arrangement. The polypeptide scaffold containing these groups must adopt a conformation that is both functionally efficient and physically strong. At first glance, the biosynthesis of polypeptides comprised of tens of thousands of individual atoms would appear to be extremely challenging. When one considers that a typical polypeptide can adopt ≥1050 distinct conformations, folding into the conformation appropriate to their biologic function would appear to be even more difficult. As described in Chapters 3 and 4, synthesis of the polypeptide backbones of proteins employs a small set of common building blocks or modules, the amino acids, joined by a common linkage, the peptide bond. Similarly, a stepwise modular pathway simplifies the folding and processing of newly synthesized polypeptides into mature proteins.
FOUR ORDERS OF THE PROTEIN STRUCTURE
The modular nature of protein synthesis and folding are embodied in the concept of orders of the protein structure: primary structure—the sequence of the amino acids in a polypeptide chain; secondary structure—the folding of short (3- to 30- residue), contiguous segments of polypeptide into geometrically ordered units; tertiary structure—the assembly of secondary structural units into larger functional units such as the mature polypeptide and its component domains; and quaternary structure—the number and types of polypeptide units of oligomeric proteins and their spatial arrangement.
SECONDARY STRUCTURE
Peptide Bonds Restrict Possible Secondary Conformations
Free rotation is possible about only two of the three covalent bonds of the polypeptide backbone: the α-carbon (Cα) to the carbonyl carbon (Co) bond, and the Cα to nitrogen bond (Figure 3–4). The partial double-bond character of the peptide bond that links Co to the α-nitrogen requires that the carbonyl carbon, carbonyl oxygen, and α-nitrogen remain coplanar, thus preventing rotation. The angle about the Cα—N bond is termed the phi (Φ) angle, and that about the Co—Cα bond the psi (Ψ) angle. For amino acids other than glycine, most combinations of phi and psi angles are disallowed because of steric hindrance (Figure 5–1). The conformations of proline are even more restricted due to the absence of free rotation of the N—Cα bond.
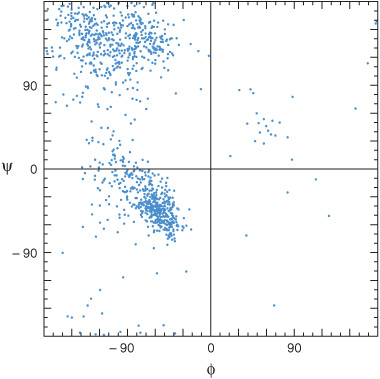
FIGURE 5–1 Ramachandran plot of the main chain phi (Φ) and psi (Ψ) angles for approximately 1000 nonglycine residues in eight proteins whose structures were solved at high resolution. The dots represent allowable combinations, and the spaces prohibited combinations, of phi and psi angles. (Reproduced, with permission, from Richardson JS: The anatomy and taxonomy of protein structures. Adv Protein Chem 1981;34:167. Copyright © 1981. Reprinted with permission from Elsevier.)
Regions of ordered secondary structure arise when a series of aminoacyl residues adopt similar phi and psi angles. Extended segments of polypeptide (eg, loops) can possess a variety of such angles. The angles that define the two most common types of secondary structure, the α helix and the β sheet, fall within the lower and upper left-hand quadrants of a Ramachandran plot, respectively (Figure 5–1).
Alpha Helix
The polypeptide backbone of an α helix is twisted by an equal amount about each α-carbon with a phi angle of approximately –57° and a psi angle of approximately –47°. A complete turn of the helix contains an average of 3.6 amino-acyl residues, and the distance it rises per turn (its pitch) is 0.54 nm (Figure 5–2). The R groups of each aminoacyl residue in an α helix face outward (Figure 5–3). Proteins contain only L-amino acids, for which a right-handed α helix is by far the more stable, and only right-handed α helices are present in proteins. Schematic diagrams of proteins represent α helices as coils or cylinders.
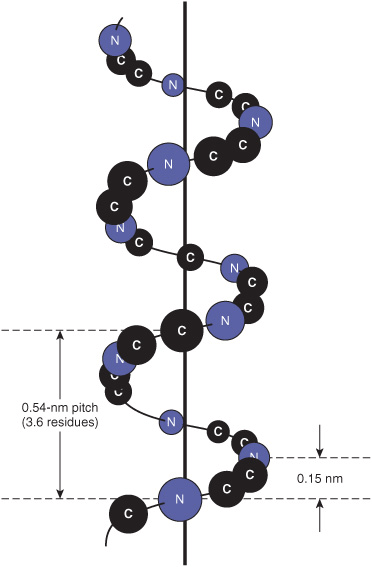
FIGURE 5–2 Orientation of the main chain atoms of a peptide about the axis of an α helix.
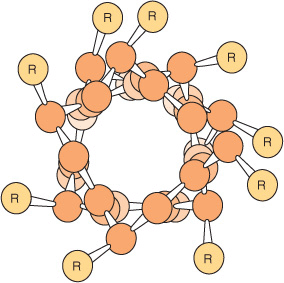
FIGURE 5–3 View down the axis of an α helix. The side chains (R) are on the outside of the helix. The van der Waals radii of the atoms are larger than shown here; hence, there is almost no free space inside the helix. (Slightly modified and reproduced, with permission, from Stryer L: Biochemistry, 3rd ed. Freeman, 1995. Copyright © 1995 W.H. Freeman and Company.)
The stability of an α helix arises primarily from hydrogen bonds formed between the oxygen of the peptide bond carbonyl and the hydrogen atom of the peptide bond nitrogen of the fourth residue down the polypeptide chain (Figure 5–4). The ability to form the maximum number of hydrogen bonds, supplemented by van der Waals interactions in the core of this tightly packed structure, provides the thermodynamic driving force for the formation of an α helix. Since the peptide bond nitrogen of proline lacks a hydrogen atom to contribute to a hydrogen bond, proline can only be stably accommodated within the first turn of an α helix. When present elsewhere, proline disrupts the conformation of the helix, producing a bend. Because of its small size, glycine also often induces bends in α helices.
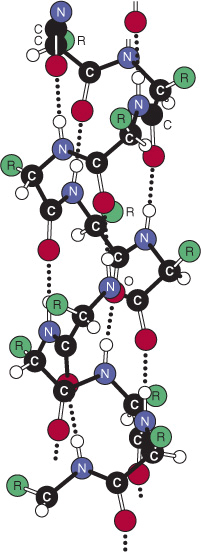
FIGURE 5–4 Hydrogen bonds (dotted lines) formed between H and O atoms stabilize a polypeptide in an α-helical conformation. (Reprinted, with permission, from Haggis GH, et al, (1964), “Introduction to Molecular Biology”. Science 146:1455-1456. Reprinted with permission from AAAS.)
Many α helices have predominantly hydrophobic R groups on one side of the axis of the helix and predominantly hydrophilic ones on the other. These amphipathic helices are well adapted to the formation of interfaces between polar and nonpolar regions such as the hydrophobic interior of a protein and its aqueous environment. Clusters of amphipathic helices can create a channel, or pore, that permits specific polar molecules to pass through hydrophobic cell membranes.
Beta Sheet
The second (hence “beta”) recognizable regular secondary structure in proteins is the β sheet. The amino acid residues of a β sheet, when viewed edge-on, form a zigzag or pleated pattern in which the R groups of adjacent residues point in opposite directions. Unlike the compact backbone of the α helix, the peptide backbone of the β sheet is highly extended. But like the a helix, β sheets derive much of their stability from hydrogen bonds between the carbonyl oxygens and amide hydrogens of peptide bonds. However, in contrast to the α helix, these bonds are formed with adjacent segments of the β sheet (Figure 5–5).
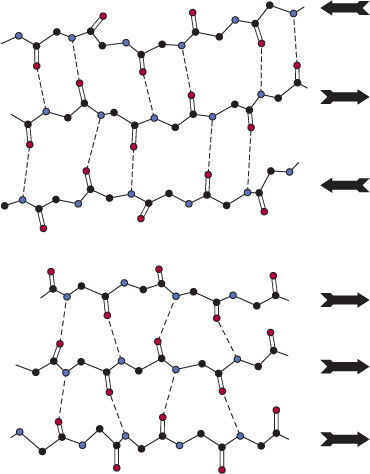
FIGURE 5–5 Spacing and bond angles of the hydrogen bonds of antiparallel and parallel pleated β sheets. Arrows indicate the direction of each strand. Hydrogen bonds are indicated by dotted lines with the participating α-nitrogen atoms (hydrogen donors) and oxygen atoms (hydrogen acceptors) shown in blue and red, respectively. Backbone carbon atoms are shown in black. For clarity in presentation, R groups and hydrogen atoms are omitted. Top: Antiparallel β sheet. Pairs of hydrogen bonds alternate between being close together and wide apart and are oriented approximately perpendicular to the polypeptide backbone. Bottom: Parallel β sheet. The hydrogen bonds are evenly spaced but slant in alternate directions.
Interacting β sheets can be arranged either to form a parallel β sheet, in which the adjacent segments of the polypeptide chain proceed in the same direction amino to carboxyl, or an antiparallel sheet, in which they proceed in opposite directions (Figure 5–5). Either configuration permits the maximum number of hydrogen bonds between segments, or strands, of the sheet. Most β sheets are not perfectly flat but tend to have a right-handed twist. Clusters of twisted strands of β sheet form the core of many globular proteins (Figure 5–6). Schematic diagrams represent β sheets as arrows that point in the amino to the carboxyl terminal direction.
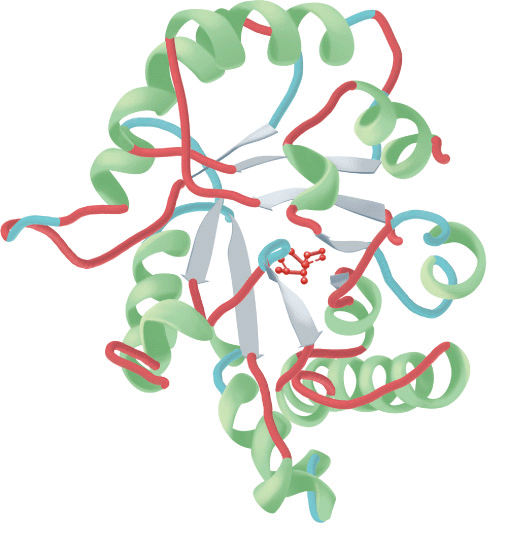
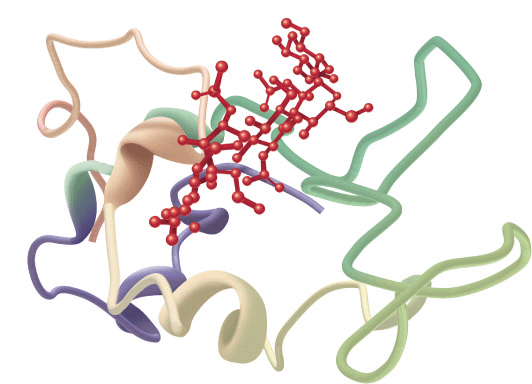
FIGURE 5–6 Examples of the tertiary structure of proteins. Top: The enzyme triose phosphate isomerase complexed with the substrate analog 2-phosphoglycerate (red). Note the elegant and symmetrical arrangement of alternating β sheets (light blue) and α helices (green), with the β sheets forming a β-barrel core surrounded by the helices. (Adapted from Protein Data Bank ID no. 1o5x.) Bottom: Lysozyme complexed with the substrate analog penta-N-acetyl chitopentaose (red). The color of the polypeptide chain is graded along the visible spectrum from purple (N-terminal) to tan (C-terminal). Notice how the concave shape of the domain forms a binding pocket for the pentasaccharide, the lack of β sheet, and the high proportion of loops and bends. (Adapted from Protein Data Bank ID no. 1sfb.)
Loops & Bends
Roughly half of the residues in a “typical” globular protein reside in α helices or β sheets, and half in loops, turns, bends, and other extended conformational features. Turns and bends refer to short segments of amino acids that join two units of the secondary structure, such as two adjacent strands of an antiparallel β sheet. A β turn involves four aminoacyl residues, in which the first residue is hydrogen-bonded to the fourth, resulting in a tight 180° turn (Figure 5–7). Proline and glycine often are present in β turns.
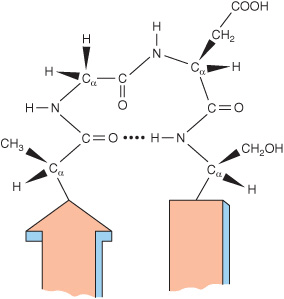
FIGURE 5–7 A β turn that links two segments of anti-parallel. β sheet. The dotted line indicates the hydrogen bond between the first and fourth amino acids of the four-residue segment Ala-Gly-Asp-Ser.
Loops are regions that contain residues beyond the minimum number necessary to connect adjacent regions of secondary structure. Irregular in conformation, loops nevertheless serve key biologic roles. For many enzymes, the loops that bridge domains responsible for binding substrates often contain aminoacyl residues that participate in catalysis. Helix-loop-helix motifs provide the oligonucleotide-binding portion of many DNA-binding proteins such as repressors and transcription factors. Structural motifs such as the helix-loop-helix motif that are intermediate in scale between secondary and tertiary structures are often termed supersec-ondary structures. Since many loops and bends reside on the surface of proteins and are thus exposed to solvent, they constitute readily accessible sites, or epitopes, for recognition and binding of antibodies.
While loops lack apparent structural regularity, many adopt a specific conformation stabilized through hydrogen bonding, salt bridges, and hydrophobic interactions with other portions of the protein. However, not all portions of proteins are necessarily ordered. Proteins may contain “disordered” regions, often at the extreme amino or carboxyl terminal, characterized by high conformational flexibility. In many instances, these disordered regions assume an ordered conformation upon binding of a ligand. This structural flexibility enables such regions to act as ligand-controlled switches that affect protein structure and function.
Tertiary & Quaternary Structure
The term “tertiary structure” refers to the entire three-dimensional conformation of a polypeptide. It indicates, in three-dimensional space, how secondary structural features—helices, sheets, bends, turns, and loops—assemble to form domains and how these domains relate spatially to one another. A domain
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


