Protein Drugs
OBJECTIVES
The reader will be able to:
- Discuss the breadth of polypeptide and protein drugs available commercially.
- State why protein drugs must generally be given parenterally.
- Compare the distribution of protein drugs within the body with that of small conventional nonprotein drugs.
- Contrast the renal and hepatic handlings of protein drugs with those of small conventional nonprotein drugs.
- State why the systemic absorption of large protein drugs is often slow and incomplete after intramuscular (i.m.) and subcutaneous (s.c.) administration.
- Describe why the lymphatic system plays a major role in the systemic absorption of protein drugs given intramuscularly or subcutaneously.
- List the primary factors affecting the rate and extent of systemic absorption of protein drugs from i.m. and s.c. sites of administration.
- Discuss the effects of renal disease on the elimination of protein drugs with molecular weights (MW) below about 30,000 g/mol.
- State why the disposition of many protein drugs is nonlinear.
- Give one example each of a pharmacokinetic and a pharmacodynamic rate-limited response seen with protein drugs.
- Discuss how nonlinear pharmacodynamics and nonlinear pharmacokinetics of protein drugs, especially antibodies, often have a mechanism in common.
PEPTIDE, POLYPEPTIDE, AND PROTEIN DRUGS
Peptide, polypeptide, and protein drugs are taking an ever larger role in the armamentarium of agents to prevent, diagnose, and treat disease. In recent years, many of the new chemical entities have been in this category. But what are peptide, polypeptide, and protein drugs?
Terminology for these drugs is not well defined, but all contain multiple amino acids that are linked via peptide bonds. They are all therefore peptides. Some have restricted the word polypeptides to peptides with 20 to 50 amino acids, with 50 as the cutoff for defining when a polypeptide becomes a protein; but there is no “official” definition. Some use a MW of 5000 g/mol as the cutoff for where proteins begin. Subsequently in this chapter, “protein” is used as an all-encompassing term for all compounds containing two or more amino acids. Antibodies are examples of proteins at the upper end of the spectrum of size with about 1300 amino acids. However, some proteins, like the largest of the lipoproteins, have more than 15,000.
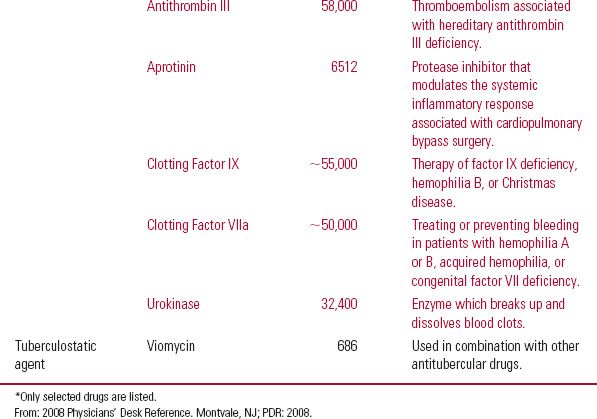
There are many types of protein drugs on the market today. Table 21-1 lists examples of nonantibody protein drugs by therapeutic class. The table demonstrates a wide range of molecular sizes and activities of the drugs in this category.
A major effort has been underway in the last decade or two to develop monoclonal antibody drugs. There are five classes of endogenous antibodies. The antibodies used as drugs belong to the immunoglobulin G (IgG) class. This group of antibodies accounts for the majority (75%–80%) of naturally occurring antibodies in the body. Collectively, they are the second most common protein in plasma (5–13 g/L); albumin is first (35–50 g/L). The general structure of IgG molecules is shown in Fig. 21-1. They are heterodimeric molecules, approximately 150,000 g/mol in size, and are composed (Fig. 21-1B) of two different kinds of polypeptide chains, heavy (~50,000 g/mol) and light (~25,000 g/mol). There are two kinds of light chains, kappa (к) and lambda (λ). Upon cleavage with the enzyme papain, the fragment–antigen binding (Fab) portion of the molecule can be separated from the Fc (fragment crystalline) portion (Fig. 21-1C). The Fab fragments contain the variable amino acid domains responsible for antibody specificity. There are literally millions of specific molecules that come under the general name IgG.
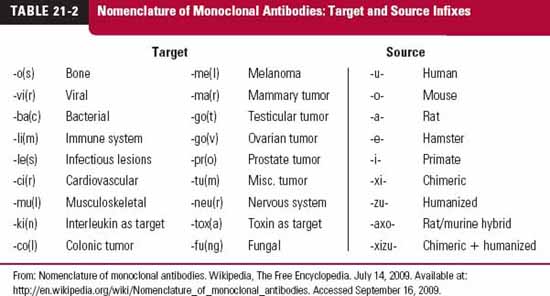
Monoclonal antibodies (mab), monospecific antibodies that are identical because they are produced by immune cells of a single type and all these cells are clones of a single parent cell, are named by the World Health Organization International Nonproprietary Name and the United States Adopted Names by a common scheme. The prefix is variable for specifying the antibody. The suffix (mab) identifies the drug as a monoclonal antibody. Infixes identifying the target system and the source of the antibody, in that order, are listed in Table 21-2. Thus, the drug adalimumab is a monoclonal antibody (-mab) of human origin (-u-), acting on the immune system (-li[m]-). To further demonstrate the naming of these drugs, selected monoclonal antibody drugs are classified in Table 21-3 based on their source, which include murine, chimeric (fusion protein), humanized, and human technologies. Also included are their therapeutic uses, their half-lives, dosing intervals, and routes of administration. Table 21-4 lists polyclonal antibodies, derived from different immune cells. Tables 21-1, 21-3, and 21-4, although quite long, are exemplary, not exhaustive. They do, however, clearly demonstrate the wide range of agents available in the category of protein drugs.
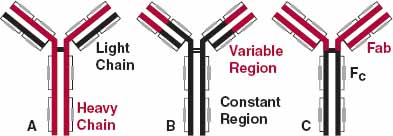
FIGURE 21-1. Antibody drugs structurally are forms of IgG. A. IgG consists of two heavy chains (colored, ~50,000 g/mol and 440 amino acids each) and two light chains (black, ~25,000 g/mol and 220 amino acids each). B. The variable portions of the heavy and light chains (colored, about 110–130 amino acids each) provide the specificity to the molecule. C. When split by papain, IgG forms two molecules of Fab (colored, fragment–antigen binding, 53,000 g/mol) and Fc (black, fragment crystalline). F(ab)2 (93,000 g/mol), the fragment formed when Fc is stripped from the rest of the molecule, is also formed. The small grey boxes denote disulfide bonds that help hold the molecule together.
It is virtually impossible to summarize the pharmacokinetic and pharmacodynamic properties of protein drugs for the category as a whole because of the wide range of compounds and activities involved. In this chapter, the general differences in the kinetic behavior of protein drugs relative to that observed with small molecules is emphasized. The kinetic and dynamic behaviors of antibody drugs are also compared to those of other protein drugs.

COMPARISON OF THE PHARMACOKINETICS OF PROTEIN DRUGS WITH THAT OF CONVENTIONAL NONPROTEIN DRUGS
As with small conventional drugs, the processes of absorption, distribution, excretion, and metabolism of protein drugs are of interest kinetically. Differences in these processes between the two kinds of drugs are subsequently examined. But first, let us consider the purity of the drugs themselves.
In Chapter 2, Fundamental Concepts and Terminology, we spent some time discussing chemical purity and analytic specificity and their importance in pharmacokinetic evaluations. Although these issues have received extensive attention for drugs that are small molecules, this has not been as easy to do with protein drugs. With protein drugs, one often uses the term heterogeneity to refer to the fact that the drug is not a pure substance but rather a mixture of substances. When MW of a protein drug is given, as in Table 21-1, it often refers to an average for the active components. In addition, the analysis of proteins in plasma or other biologic fluids is often based on immunoassays, which show some cross-reactivity with similar proteins. What is being measured is always of concern. With these caveats in mind, let us examine the general pharmacokinetic differences between protein drugs and small conventional ones.
ABSORPTION
In contrast to small conventional drugs, systemic absorption is a major problem with protein drugs. These kinds of drugs generally cannot be taken orally because of their instability in the gastrointestinal tract and great difficulty in permeating the intestinal epithelium. By this route, they are, simply put, a source of amino acids for protein synthesis within the body. As a consequence, their oral bioavailabilities are usually extremely low and erratic. They generally must be given parenterally. However, even when given subcutaneously or intramuscularly, common routes of their administration, their systemic bioavailabilities are often low and variable, an area explored further later in the chapter. To circumvent the lack of absorption after oral administration, some proteins are only used locally or are given for their systemic effect by less conventional methods as shown in Table 21-5. In the case of the leuprolide acetate polymeric matrix formulations and its implant, as discussed in Chapter 7, Absorption, the intent is not just systemic delivery, but to prolong its systemic input. Indeed, considerable effort has been, and will continue to be, placed on developing pharmaceutical systems to improve systemic delivery of protein drugs.
DISTRIBUTION
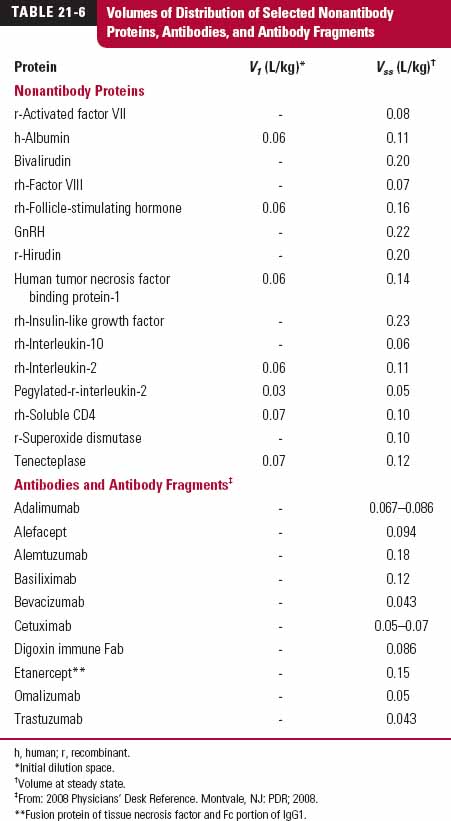
With few exceptions, protein drugs are hydrophilic molecules that have difficulty entering cells of the body. They therefore tend to have volumes of distribution that lie between that of plasma (0.04 L/kg) and the extracellular space (0.23 L/kg), as shown in Table 21-6 for both nonantibody and antibody proteins. For those that enter the extra-cellular matrix of tissues, they often do so slowly, particularly large ones, because they have difficulty permeating the blood capillary endothelium (as discussed later in this chapter). One exception to this generalization of a small volume of distribution is the immunosuppressive drug, cyclosporine (not in Table 21-6), a cyclic undecapeptide with a MW of 1202 g/mol and a volume of distribution of 3 to 5 L/kg. This unusual peptide, having no charge at physiologic pH, is highly lipophilic with an noctanol/water partition coefficient of about 1000 and a high affinity for tissues, especially adipose tissue.
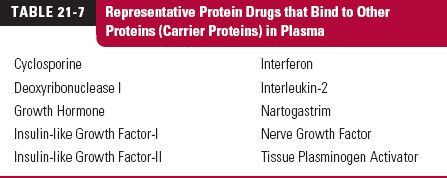
Many protein drugs that are endogenous substances have carrier proteins within the body that may improve their stability as well as aid in the delivery to the site of action at a higher rate than would occur if there were no such binding. Examples of such protein drugs with carrier proteins are given in Table 21-7 (page 643). Some carriers also help to stabilize the protein. An example is that of human growth hormone, Table 21-8 (page 643). When given with human growth hormone–binding protein, clearance of the drug and its volume of distribution are greatly reduced in the rat, an animal that has neither the human binding protein nor human growth hormone.

Binding sometimes occurs within the extracellular space or on cell membranes of tissues. As with small drugs, the volume of distribution of a protein depends on the relative binding within tissues and plasma, as well as the aqueous space into which the protein gains access. Furthermore, for large proteins, the steady-state unbound concentration of protein in interstitial fluid is much lower than that in blood. This is different from our expectation for most small molecule drugs, where in many cases diffusion is the primary mechanism responsible for drug distribution. Protein concentrations are lower in the interstitial fluid and vary from tissue to tissue because of differences in the efficiency of convective uptake, capillary permeability, lymph flow, and tissue metabolism.
In many cases, it may be impossible to infer accurate distribution information from plasma data alone. Common methods applied for analysis of Vss and V assume that all elimination occurs at sites that are in rapid equilibrium with plasma. This assumption is valid in most cases for small molecules, as most elimination occurs in the liver and kidney, organs that are very highly perfused. Many proteins, however, may be eliminated from sites that are poorly perfused, and application of mammillary models (Chapter 19, Distribution Kinetics) may lead to significant underestimations of the actual extent of drug distribution.

RENAL EXCRETION
No matter the size, protein drugs, which are usually highly hydrophilic, generally have low renal clearances (when defined as rate of urinary excretion relative to the plasma concentration). However, this does not mean that many are not extensively eliminated in the kidneys. The low renal clearance of protein drugs arises from one of two properties: their size, proteins larger than about 60,000 g/mol are not materially filtered, or from their metabolism (or degradation) in the proximal tubule after their filtration. Because of extensive metabolism of smaller proteins, it is better to use the terms renal handling or renal processing, instead of renal excretion to describe the elimination of protein drugs in the kidneys.
The key to understanding the loss of protein drugs in the kidneys, as viewed from plasma, is glomerular filtration. Although 60,000 g/mol has been given as a rough cutoff for filtration, molecular size is not the only factor determining the extent of filtration. Table 21-9 lists the glomerular sieving coefficients (concentration in glomerular filtrate relative to that unbound in plasma) of selected proteins. It is apparent from the data for horseradish peroxidases that charge also plays a role. This is reinforced by studies with dextrans (branched polysaccharides made with glucose molecules) prepared with and without charges on them (Fig. 21-2). Anionic dextrans undergo filtration much less readily than cationic ones at the same molecular size, measured here by effective molecular radius (crudely, MW = [1.14 × molecular radius]3). Albumin, a protein with a MW of 69,000 g/mol, has a negative charge and thus has a sieving coefficient less than that expected had it been neutral or cationic in nature. Shape of the molecule and rigidity also play a role as does binding to other proteins within plasma. Those proteins that are bound to large carrier proteins have reduced glomerular filtration as one would expect.
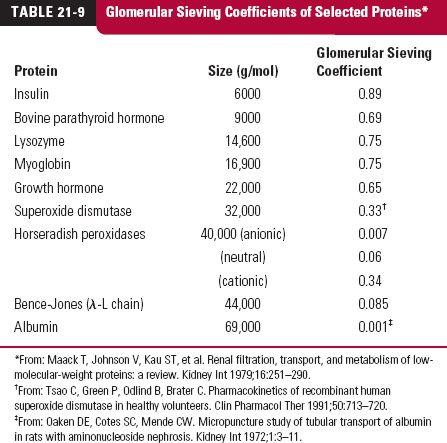
FIGURE 21-2. Glomerular sieving coefficients of dextran derivatives reflect the influence of size and charge of molecules. Cationic molecules tend to be much more readily filtered than neutral ones which, in turn, are more readily filtered than anionic ones. Added is a value for albumin ( ), a molecule with a net negative 52 charges at physiologic pH. (From: Arendshorst WJ, Navar LG. Renal circulation and glomerular hemodynamics. In: Schrier RW, ed. Diseases of the Kidney and Urinary Tract. 8th ed. Philadelphia: Lippincott Williams & Wilkins; 2006.)
), a molecule with a net negative 52 charges at physiologic pH. (From: Arendshorst WJ, Navar LG. Renal circulation and glomerular hemodynamics. In: Schrier RW, ed. Diseases of the Kidney and Urinary Tract. 8th ed. Philadelphia: Lippincott Williams & Wilkins; 2006.)
As shown in Fig. 21-3, low MW proteins are often extensively metabolized by enzymes located in the brush border of the lumen, whereas higher MW proteins are transported into the proximal tubular cells by endocytosis and metabolized (degraded) there by lysosomal enzymes. Generally, catabolism of proteins continues until the constituent amino acids are formed, although some are only partially degraded. For small proteins that are metabolized on the brush border, the amino acids and fragments are largely conserved by trans porters that reabsorb them back into the vasculature. Some peptides and small proteins undergo postglomerular and peritubular transport and metabolism. This is analogous to secretion of small molecules, but little or none of the protein drug may appear in the urine. Although quantitative information is often lacking, for many protein drugs with a molecular size less than 30,000 g/mol, the kidney is a, if not the, major organ of elimination.
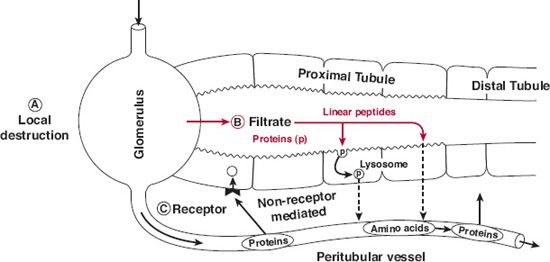
FIGURE 21-3. Pathways of protein elimination in the kidney. A. Local degradation by the glomerulus represents a minor site of elimination for small peptide hormones such as angiotensin, bradykinin, atrial natriuretic peptide, and possibly calcitonin. B. Glomerular filtration and tubular absorption is the major route for proteins and peptides. Proteins and complex peptides require internalization prior to degradation. Small linear peptides are hydrolyzed at the luminal membrane. C. Postglomerular peritubular elimination constitutes an important though lesser route of elimination for some protein hormones. Their basolateral uptake is largely receptor mediated and is followed by intracellular degradation. (From: Rabkin R, Dahl DO. Renal uptake and disposal of proteins and peptides. In: Audus KL, Raub TJ, eds. Biological Barriers to Protein Delivery. New York: Plenum Press; 1993.)
METABOLISM
Proteins are metabolized by proteolytic enzymes, which ultimately break the substances down to their constituent amino acids. These enzymes are ubiquitous, some within cells, some on cell surfaces, and some in extracellular fluids in essentially all tissues of the body. Virtually all protein drugs are eliminated by metabolism; exceptions appear to be restricted to a few small peptides, which are primarily eliminated by renal excretion. In general, however, metabolism predominates, but where the metabolism occurs varies from one protein to another. Size of the molecule is particularly important. Indeed, two issues seem to be overriding in determining the location and the rate of metabolism of proteins, namely, substrate specificity/reactivity, and accessibility to the metabolizing enzymes. Glycosylation, addition of glycan (carbohydrate) to a protein and pegylation, attaching one or more chains of polyethylene glycol, usually increase the half-life of a protein by either making it a poorer substrate for proteolysis or decreasing its access to the metabolizing enzymes, or both. Certainly, they have the potential to reduce glomerular filtration and renal metabolism of those polypeptides and proteins that become larger than about 30,000 g/mol. Provided that activity is retained, this procedure has therefore become a common one to try to improve therapy with certain protein drugs.
For antibody drugs, being very large, there is no glomerular filtration in the kidneys, and the liver appears to not contribute much to their metabolism either. The majority of antibody elimination occurs following fluid-phase endocytosis, by cells at sites throughout the body, followed by intracellular catabolism. As mentioned previously, generally, the clearances of antibodies are extremely low when only this process is involved in elimination. This relative stability may be caused, at least in part, by binding to the Brambell receptor (FcRn), which protects the antibody by preventing its uptake and degradation by the acidic lysosomes within the cell. The complex is eventually returned to the cell surface, where it rapidly dissociates to liberate the antibody.
PHARMACODYNAMICS
PHARMACODYNAMICS OF NONANTIBODY PROTEIN DRUGS
Nonantibody proteins have a wide range of pharmacologic effects (see Tables 21-1 and 21-3
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


