22
Prostaglandins, NSAIDs, and Pharmacotherapy of Gout
This chapter will be most useful after having a basic understanding of the material in Chapter 33, Lipid-Derived Autacoids: Eicosanoids and Platelet-Activating Factor, and Chapter 34, Anti-inflammatory, Antipyretic, and Analgesic Agents: Pharmacotherapy of Gout in Goodman & Gilman’s The Pharmacological Basis of Therapeutics, 12th Edition. In addition to the material presented here, the 12th Edition contains:
• Table 33-1 Eicosanoid Receptors that lists the major classes of eicosanoid receptors and their signaling characteristics
• The molecular structures of lipid-derived autacoids, nonsteroidal anti-inflammatory drugs (NSAIDs), and drugs used to treat gout
LEARNING OBJECTIVES
 Understand the mechanisms of action of drugs used as prostaglandin agonists.
Understand the mechanisms of action of drugs used as prostaglandin agonists.
 Understand the mechanisms of action of the NSAIDs.
Understand the mechanisms of action of the NSAIDs.
 Understand the mechanisms of action of the drugs used in the pharmacotherapy of gout.
Understand the mechanisms of action of the drugs used in the pharmacotherapy of gout.
 Know the untoward effects of prostaglandin agonists, NSAIDs, and the drugs used in the pharmacotherapy of gout.
Know the untoward effects of prostaglandin agonists, NSAIDs, and the drugs used in the pharmacotherapy of gout.
 Know the therapeutic utility of prostaglandin agonists, NSAIDs, and the drugs used in the pharmacotherapy of gout.
Know the therapeutic utility of prostaglandin agonists, NSAIDs, and the drugs used in the pharmacotherapy of gout.
 Know which drugs can be used in combination, and those that should not be used concomitantly to treat inflammatory disorders, fever, pain, arthritis, and gout.
Know which drugs can be used in combination, and those that should not be used concomitantly to treat inflammatory disorders, fever, pain, arthritis, and gout.
DRUGS INCLUDED IN THIS CHAPTER
• Acetaminophen (TYLENOL, others)
• Allopurinol (ZYLOPRIM, ALOPRIM, others)
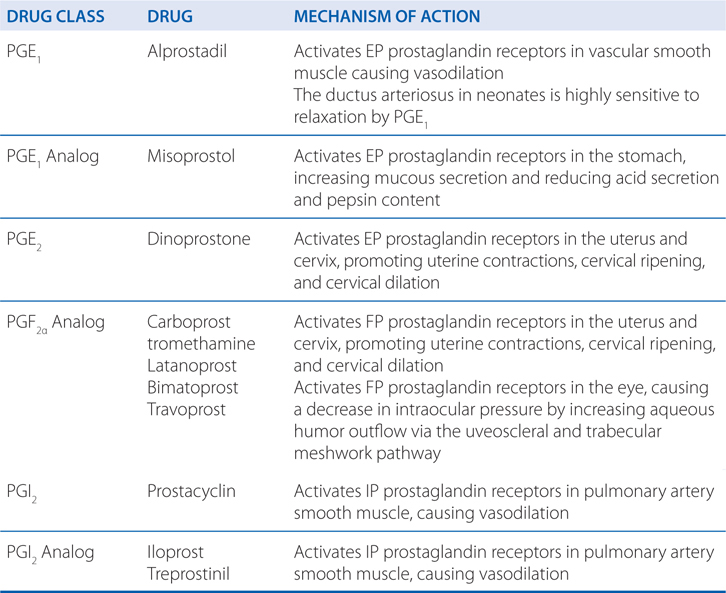
• Alprostadil (CAVERJECT, EDEX, MUSE, PROSTIN VR PEDIATRIC)
• Aspirin (acetylsalicylic acid, ASA)
• Balsalazide (COLAZOL, others)
• Bimatoprost (LUMIGAN)
• Bromfenac (XIBROM)
• Carboprost tromethamine (HEMABATE)
• Celecoxib (CELEBREX)
• Colchicine
• Diclofenac (CATAFLAM, ZIPSOR, FLECTOR, VOLTAREN, SOLAREZE, CAMBIA, others)
• Diflunisal
• Dinoprostone (CERVIDIL, PREPIDIL)
• Etodolac (LODINE)
• Febuxostat (ULORIC)
• Fenoprofen (NALFON 200, others)
• Flurbiprofen (ANSAID, OCUFEN, others)
• Ibuprofen (ADVIL, MOTRIN, NEOPROFEN, CALDOLOR, others)
• Iloprost (VENTAVIS)
• Indomethacin (INDOCIN, others)
• Ketoprofen
• Ketorolac (ACULAR, others)
• Latanoprost (XALATAN)
• Meclofenamate (MECLOMEN)
• Mefenamic acid (PONSTEL)
• Meloxicam (MOBIC, others)
• Mesalamine (ASACOL, LIALDA, APRISO, DIPENTUM, CANASA)
• Misoprostol (CYTOTEC)
• Nabumetone (RELAFEN)
• Naproxen (ALEVE, NAPROSYN, others)
• Nepafenac (NEVANAC)
• Olsalazine
• Oxaprozin (DAYPRO)
• Piroxicam (FELDENE, others)
• Probenecid (PROBALAN)
• Prostacyclin (epoprostenol, FLOLAN)
• Rasburicase (ELITEK)
• Salicylate (DOAN’S, MOMENTUM, others)
• Salsalate
• Sulfasalazine (AZULFIDINE, others)
• Sulindac (CLINORIL, others)
• Tolmetin (TOLECTIN, others)
• Travoprost (TRAVATAN)
• Treprostinil (REMODULIN)
MECHANISMS OF ACTION OF PROSTAGLANDINS AND THEIR ANALOGS
MECHANISMS OF ACTION OF NSAIDs

THE INFLAMMATORY RESPONSE
• Inflammation can be evoked by a wide variety of noxious agents (eg, infections, antibodies, physical injuries).
• The classic inflammatory symptoms include calor (warmth), dolor (pain), rubor (redness), and tumor (swelling).
• Many mediators are involved in the inflammatory response:
▶ Histamine (see Chapter 21)
▶ Bradykinin (see Chapter 21)
▶ 5-hydroxytryptamine (5-HT, serotonin)
▶ Prostanoids produced by cyclooxygenase-1 (COX-1) and cyclooxygenase-2 (COX-2) (see Figure 22-1), in particular, PGE2 and PGI2
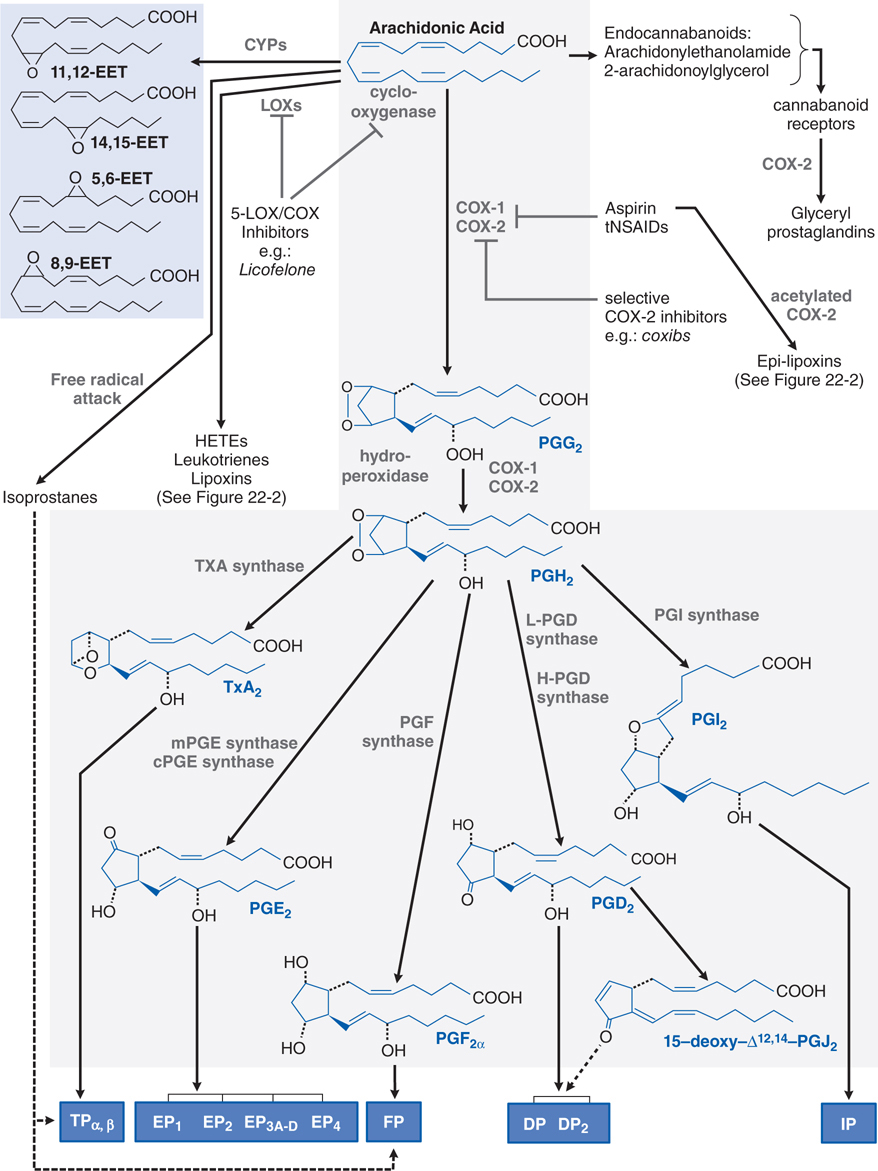
FIGURE 22-1 Metabolism of arachidonic acid (AA). The cyclooxygenase (COX) pathway is highlighted in gray. The lipoxygenase (LOX) pathways are expanded in Figure 22-2. Cyclic endoperoxides (PGG2 and PGH2) arise from the sequential COX and hydroperoxidase actions of COX-1 or COX-2 on AA released from membrane phospholipids. Subsequent products are generated by tissue-specific synthases and transduce their effects via membrane-bound receptors (blue boxes). Dashed lines indicate putative ligand–receptor interactions. Epoxyeicosatrienoic acids (EETs; shaded in blue) and isoprostanes are generated via CYP activity and nonenzymatic free radical attack, respectively. COX-2 can use modified arachidonoylglycerol, an endocannabinoid, to generate the glyceryl prostaglandins. Aspirin and tNSAIDs are nonselective inhibitors of COX-1 and COX-2, but do not affect LOX activity. Epi-lipoxins are generated by COX-2 following its acetylation by aspirin (see Figure 22-2). Dual 5-LOX–COX inhibitors interfere with both pathways.
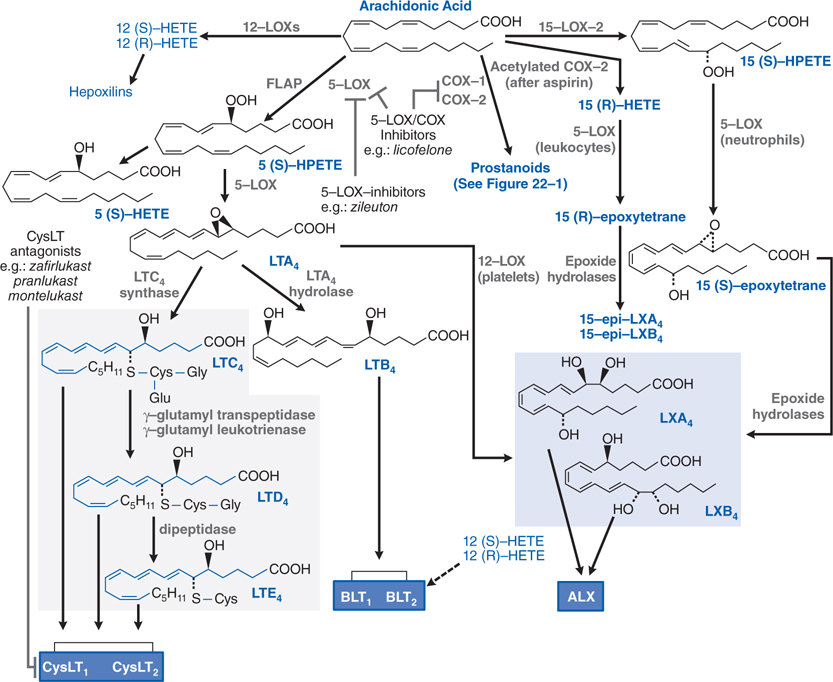
FIGURE 22-2 Lipoxygenase pathways of arachidonic acid metabolism. 5-LOX–activating protein (FLAP) presents arachidonic acid to 5-LOX, leading to the generation of the LTs. CysLTs are shaded in gray. Lipoxins (shaded in blue) are products of cellular interaction via a 5-LOX–12-LOX pathway or via a 15-LOX–5-LOX pathway. Biological effects are transduced via membrane-bound receptors dark (blue boxes). The dashed line indicates putative ligand–receptor interactions. Zileuton inhibits 5-LOX but not the COX pathways (expanded in Figure 22-1). Dual 5-LOX–COX inhibitors interfere with both pathways. CysLT antagonists prevent activation of the CysLT1 receptor.
▶ Leukotrienes (LTs; see Figure 22-2)
▶ Platelet-activating factor (PAF; see Figure 22-3)
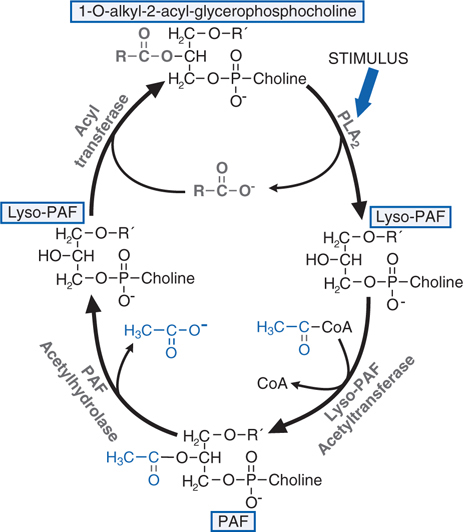
FIGURE 22-3 Synthesis and degradation of platelet-activating factor (PAF). RCOO– is a mixture of fatty acids but is enriched in arachidonic acid that may be metabolized to eicosanoids. CoA, coenzyme A.
▶ Other soluble mediators including complement factor C5a, proinflammatory cytokines (TNF, IL-1, IL-2, IL-6, IL-8; see Chapters 23 and 25), and growth factors (GM-CSF; see Chapter 25)
• COX-2, which is rapidly induced during inflammation, is the major source of proinflammatory prostanoids.
• COX-1 plays a dominant role in producing proinflammatory prostanoids during the initial phase of an acute inflammatory response.
TABLE 22-1 Classification and Comparison of Nonsteroidal Analgesics
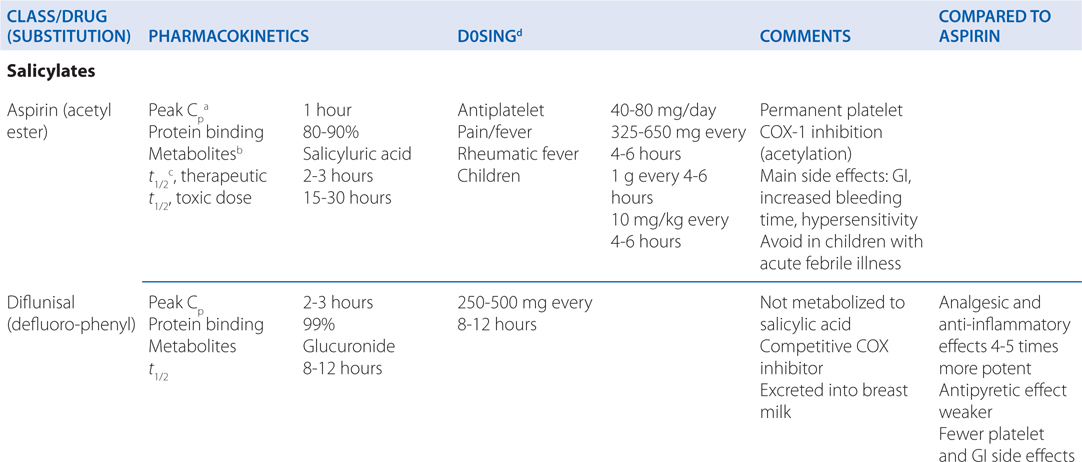
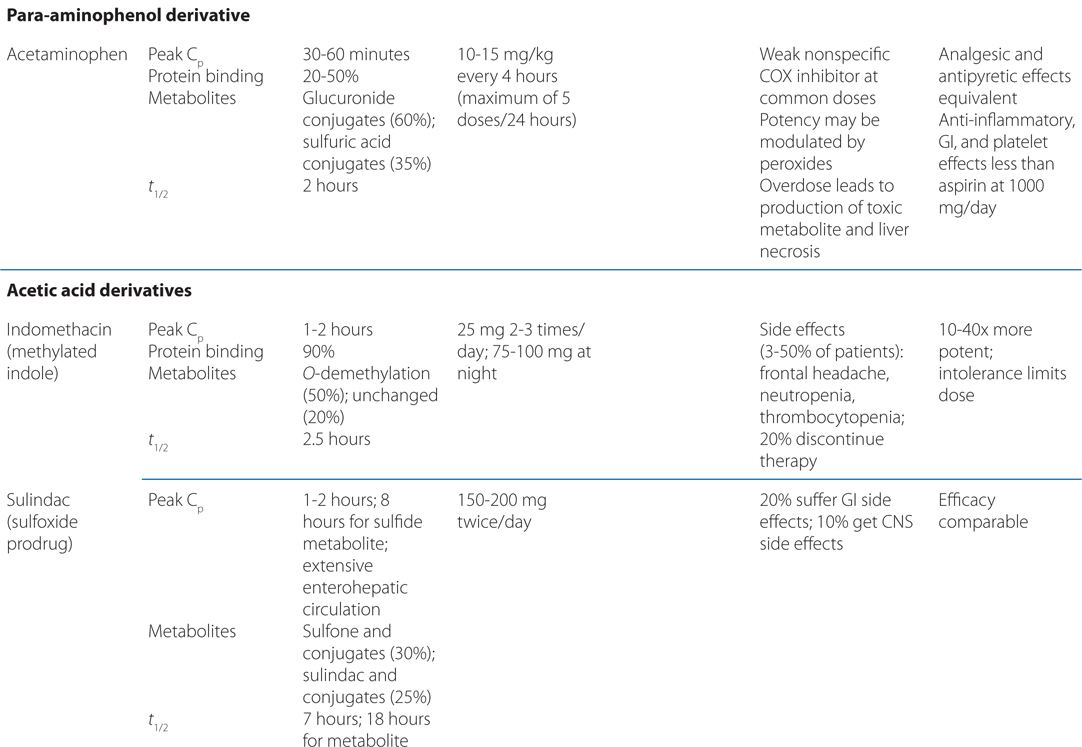
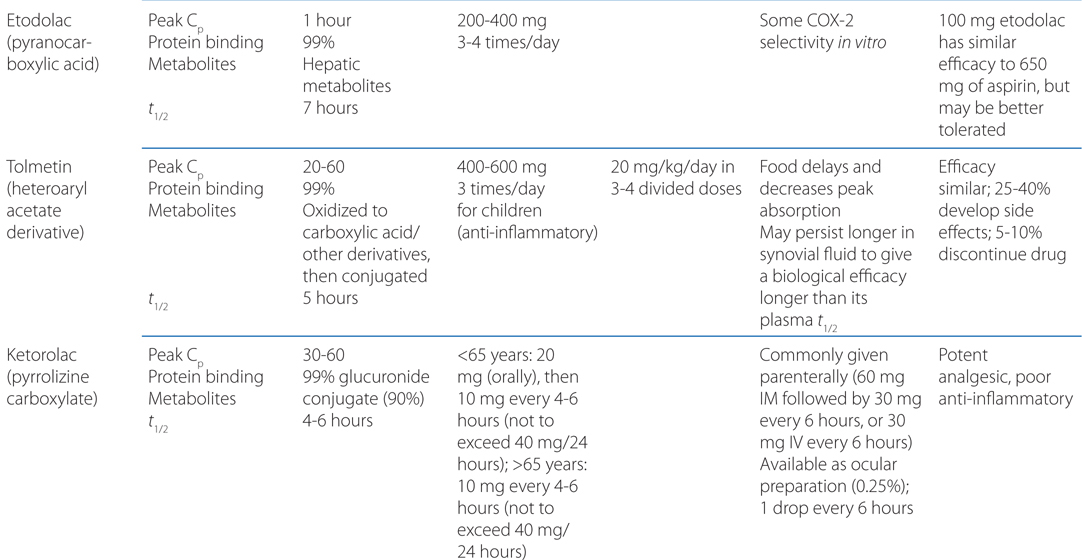
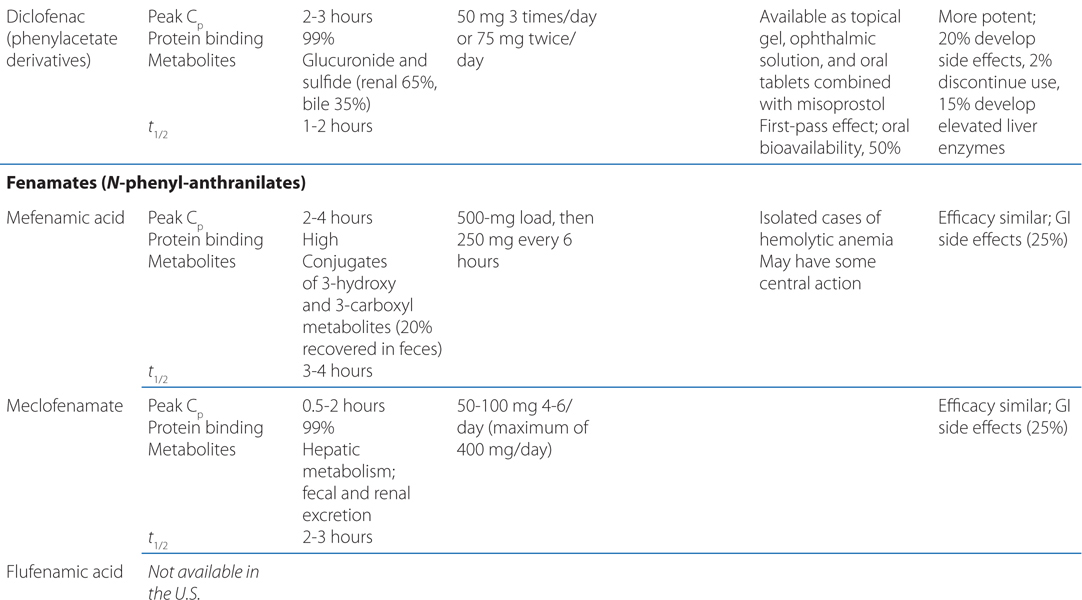
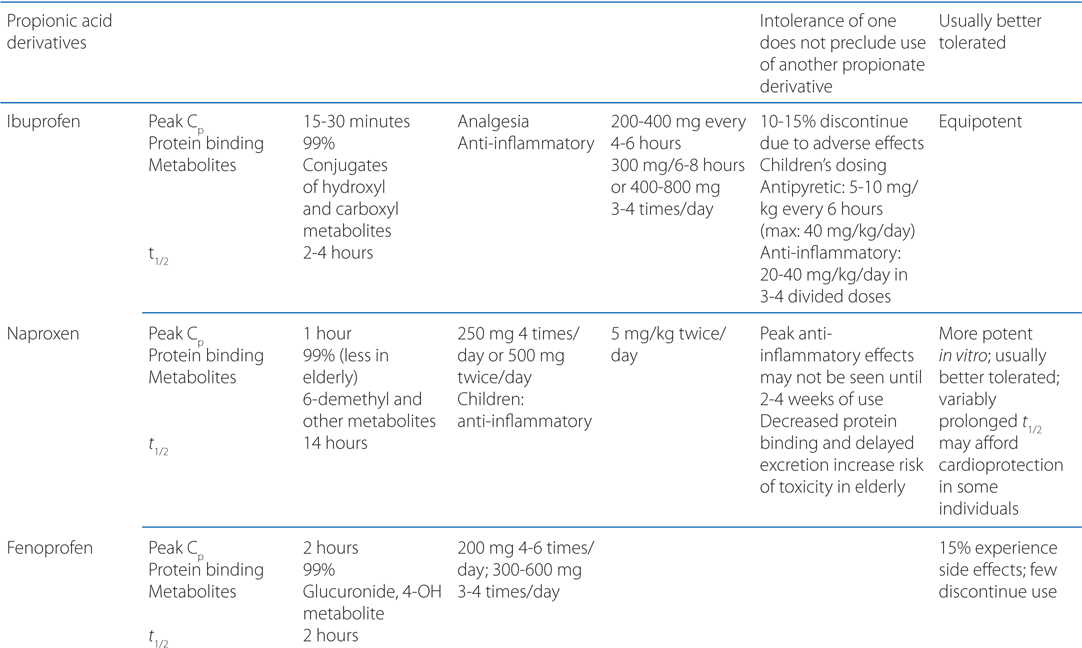
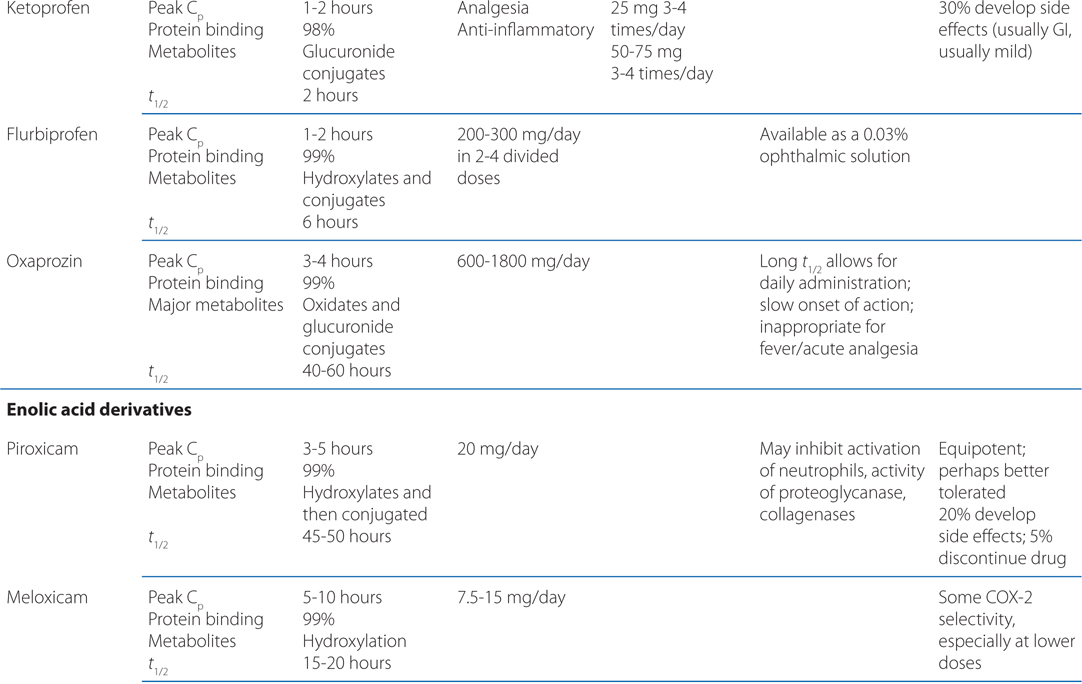
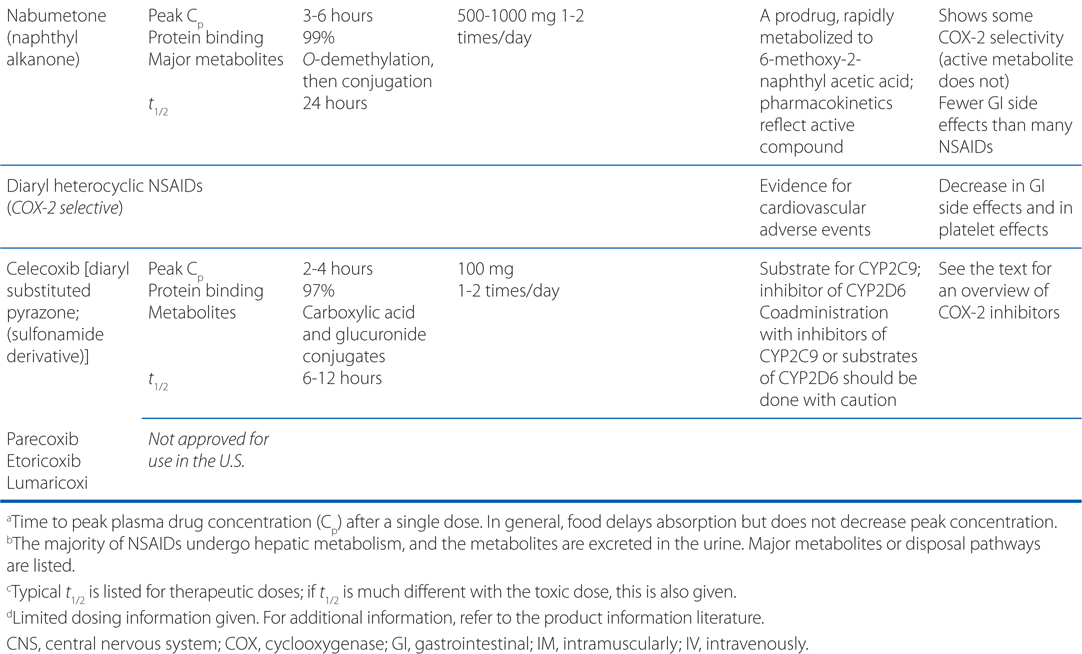
• All NSAIDs, including COX-2 Inhibitors, are anti-inflammatory, analgesic, and antipyretic, except for acetaminophen, which is largely devoid of anti-inflammatory activity.
▶ Used as anti-inflammatory agents in the treatment of musculoskeletal disorders (the primary clinical application of NSAIDs)
■ Rheumatoid arthritis
■ Osteoarthritis
■ Ankylosing spondylitis
■ Gout
▶ Used as analgesics to treat pain of low to moderate intensity
■ NSAIDs lack the unwanted adverse CNS effects of opiates (respiratory depression and development of physical dependence).
■ Coadministration of NSAIDs with opioids can reduce the dose of opiate and the likelihood of adverse CNS effects.
■ NSAIDs are particularly effective in treating dental pain, postoperative pain, pain arising from inflammation, and menstrual cramps (the latter due to release of prostaglandins from the endometrium).
■ NSAIDs are commonly used as first-line agents in treating migraine attacks and can be combined with second-line agents such as the triptans, or with antiemetics for relief of nausea.
■ NSAIDs lack efficacy in treating neuropathic pain or pain arising from the hollow viscera.
▶ Can reduce fever in most situations, but antipyretic effects may obscure the course of illness
■ Antipyretic therapy is reserved for patients with deleterious fever and those who experience relief when fever is lowered.
• Used in neonates to close inappropriately patent ductus arteriosus, which is very sensitive to the dilating effects of PGE1.
• Low-dose aspirin reduces the risk of serious cardiovascular events in high-risk patients, probably by irreversibly inhibiting thromboxane A2 (TxA2) production by platelets (see Chapter 19).
A 24-year-old woman experiences severe menstrual cramping each month. The cramping and pain are relieved when she takes ibuprofen.
a. What is causing this patient’s menstrual cramping?
The release of PGs by the endometrium during menstruation may cause severe cramps and other symptoms of primary dysmenorrhea.
b. What is the mechanism of action of ibuprofen in relieving this patient’s pain and cramping?
Ibuprofen and other NSAIDs are effective in treating menstrual cramping because of their ability to inhibit synthesis of PGs by the endometrium through their inhibitory effects on COX-1 and COX-2. The selective COX-2 inhibitors also are efficacious in this condition.
c. If this patient becomes pregnant, are there any hazards to her or the fetus in using NSAIDs for relief of headaches and musculoskeletal pains?
The use of NSAIDs and aspirin late in pregnancy may increase the risk of postpartum hemorrhage. Therefore, pregnancy, especially close to term, is a relative contraindication to the use of all NSAIDs. In addition, their use must be weighed against potential fetal risk, even in cases of premature labor, and especially in cases of pregnancy-induced hypertension. NSAID use is associated with closure of the ductus arteriosus and impaired fetal circulation in utero, particularly in fetuses older than 32 weeks’ gestation.
MECHANISMS OF GASTRIC DAMAGE BY NSAIDs
• Inhibition of COX-1 in gastric epithelial cells depresses mucosal cytoprotective PGs, especially PGE2 and PGI2.
▶ COX-2 inhibition in gastric epithelial cells may also contribute to some loss of cytoprotective PGs.
• Local irritation of gastric mucosa by orally administered NSAIDs.
• Enhanced generation of lipoxygenase (LOX) products (eg, LTs; see Figure 22-2) may contribute to ulcerogenicity in patients treated with NSAIDs.
A 64-year-old man who suffered a myocardial infarction 2 years earlier is taking 80 mg of aspirin each day on the recommendation of his family physician. His doctor has advised him that this will reduce his chances of another myocardial infarction or stroke.
a. What is the mechanism of aspirin’s cardioprotective effect in this patient?
This effect is due to irreversible acetylation of platelet COX-1, which permanently and completely suppresses platelet thromboxane A2 (TxA2) formation, with the consequent inhibition of platelet function (see Figure 19-1). Platelets, which being anucleate, have a markedly limited capacity for protein synthesis. Thus, the consequences of inhibition of platelet COX-1 (COX-2 is not expressed in mature platelets) last for the lifetime of the platelet. Inhibition of platelet COX-1–dependent TxA2 formation, therefore, is cumulative with repeated doses of aspirin (at least as low as 30 mg/d) and takes ~8 to 12 days—the platelet turnover time—to recover fully once therapy has been stopped. Low-dose aspirin therapy does not significantly impair synthesis of PGI2 by vascular endothelium, in part because of the ability of endothelial cells to continuously express COX-1 and COX-2. Thus, daily low-dose aspirin blocks the formation of TxA2, which promotes platelet aggregation, while leaving intact the synthesis of PGI2, which inhibits platelet function (see Figure 19-1).
Aspirin reduces the risk of serious vascular events in high-risk patients (eg, those with previous myocardial infarction) by 20 to 25%. Low-dose (<100 mg/d) aspirin, which is relatively (but not exclusively) selective for COX-1, is as effective as higher doses (eg, 325 mg/d).
CARDIOVASCULAR HAZARD WITH COX-2–SELECTIVE NSAIDs
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


