, Mohammed Al-Rubaie2, Stuart Walker3 and Sam Salek4
(1)
Regulatory Affairs Consultant Executive Directive, Kuwait Advancement for Conference and Exhibition Management, Kuwait, Kuwait
(2)
Ministry of Health, Directorate General of Pharmaceutical Affairs and Drug Control, Muscat, Oman
(3)
Founder of Centre for Innovation in Regulatory Science, London, UK
(4)
Department of Pharmacy School Life & Medical Sciences, University of Hertfordshire, Hatfield, UK
Introduction
The Centralised Procedure is designed to improve the operation of the ‘single market’ for medicinal products, the avoidance of duplication of scientific evaluation and the reduction of the administrative burden. Apart from these, product safety enhancement and harmonisation are also important for this region. However, the situation in the Gulf centralised registration procedure is complex because each state has its own government, constitution, legislation, regulatory requirements and healthcare systems, as well as differences in history and medical practices.
Today, regulatory authorities are thinking in terms of global and regional themes, for example, the European Medicines Agency (EMA), the African Medicines Regulatory Harmonization (AMRH) initiative and the International Conference on Harmonisation (ICH) for Europe, Japan and USA. However, such efforts are not risk-free due to the influence of regional groups in pursuing harmonisation of drug registration. In addition, more stringent registration requirements may displace substandard medicines from the market. The Ministers of Health in most of the Gulf states have expressed their doubts about the ability to secure the expertise required for registration whilst at the same time there is the fear of losing their sovereignty to the centralised authority (Rahman 2002). The successful development and registration of a medicinal product requires effective dialogue between the pharmaceutical industry and the health authorities. For the pharmaceutical industry, good communication with the health authorities is essential during the entire lifecycle of a medicinal product, which includes the registration procedure and the subsequent maintenance phase (Heidenreich 2004). The main goal of both the pharmaceutical industry and the health authorities is to establish a positive benefit-risk ratio and an appropriate benefit-risk management system for medicinal products. Pharmaceutical companies should contact the health authorities at an early stage in the development in order to achieve this goal. Early and frequent dialogue between these two stakeholders can build a good relationship between the future assessors of the marketing authorisation applications and the responsible managers of the company. This may rule out problems and issues with the development of the product and thus reduce the risk of an application being rejected. The result will be more rapid access to essential priority medicines, including important new treatment options such as generic fixed-dose combinations of assured quality and affordable prices by the communities and patients in the Gulf states. National drug regulatory authorities need to be better equipped to register medicines in a cost-effective and timely manner, and this will lead to increased efficiencies across GCC countries. Capacity building will enhance the quality of the registration decision, whilst streamlined processes and improved information management lead to effective medicines control. As a result, pharmaceutical regulatory authorities may enjoy substantial savings and reach more patients with generic equivalents in the context of high-volume and high-value essential medicines. Beyond this, governments could enjoy additional savings via pooled procurement, which is only possible when the same medicine is registered across all participating countries.
Similarities and Differences Between the Experiences of the Drug Regulatory Authorities and the Pharmaceutical Companies
At the introduction of the GCC-DR system in 1999, government officials were concerned about losing sovereignty to the centralised authority (Hashan 2005). The central registration system has faced criticism for some aspects and approval for others from both the member states of the centralised authority and the pharmaceutical companies dealing with the system. An interesting initiative which contributed to improvement of the GCC-CP was the appointment of ‘Cameron McKenna and Andersen Consulting’ to audit the EMA’s centralised and decentralised drug registration procedures. An EMA audit survey mandated a review of that agency’s activity within 6 years of its inception.
The report of this survey, namely, the ‘Evaluation of Community Producers for the Authorisation of Medicinal Products’, was published in October 2000. It was based on interviews with and/or questionnaire responses from trade associations, national drug registration authorities, professional and patient associations, national ministries with interests in drug approval and pharmaceutical companies. The audit report contained the following summary of survey responses:
The centralised and decentralised procedures are both perceived to have contributed to the creation of a harmonised community market in medicinal products.
There is some criticism of particular aspects of both systems. However, in general, the two systems provide a strong foundation for future progress to a harmonised and efficient regulatory environment. Because of their different attributes, there is a strong desire on the part of both applicants for marketing authorisations and the competent regulatory authorities to maintain the parallel systems.
The EMA survey examined many aspects of both the centralised and mutual recognition procedures. The auditors did not limit their research to the EMA’s activities, but they investigated member states’ response to EMA decisions. This survey examined the level of satisfaction with the Centralised Procedure. Of the 32 companies that had obtained marketing authorisations by this route, 88 % were satisfied with the system and 3 % was very satisfied. The member states regulatory authorities of EMA were more enthusiastic about the Centralised Procedure with 73 % being satisfied whilst 20 % were very satisfied with its operation. This concurs with the experience of the regulatory authorities and pharmaceutical companies in the Gulf region.
A well-coordinated, reliable centralised regulatory framework effectively reduces the administrative burden and duplication of scientific evaluations by participating member states. The companies believed that if this procedure was streamlined, was faster, and transparent, it could be the system of choice. As the documents for a medicinal product are submitted as a common single registration, this results in cost saving and an efficient follow-up opportunity in all the seven Gulf states. At the beginning of 2014, the GCC-DR initiated the CTD electronic submission (as NeeS [Non-eCTD electronic Submissions] format), which is currently implemented by internationally recognised agencies. However, the GCC countries are still building their internal capacities in order to implement eCTD format. This approach is useful in that it assists the pharmaceutical companies in understanding the rules of the submission process and thus helps both the industry and the authority to make better decisions (TGA 2009). The findings from this study highlight the 10 years achievement of the Centralised Procedure and its future potential as identified by the health authorities as well as the pharmaceutical companies. The pharmaceutical companies shed some light on the advantages and disadvantages of the centralised system which were similar to the views of the national regulatory authorities of the member states, although in some ways they were different. Both the regulatory authorities and the pharmaceutical companies agreed that the Centralised Procedure is an effective system for authorising the marketing of medicinal products in all of the GCC countries in one procedure and is the way forward, but there is room for improvement in the procedure and the follow-ups.
With regard to submission preference, the majority of the pharmaceutical companies, which reported their views, preferred the review at a national level mainly due to their experience with the national regulatory authorities, therefore making it easier to gain marketing authorisation. The long duration of the CP process contributes to the preference for the national submission as well as the fear of rejection by the centralised authority. The preference of the pharmaceutical companies to apply to national authorities is confirmed by the responses of the national regulatory authorities which stated that the number of national applications was greater than those submitted to the centralised authority adding to the workload of the national regulatory authorities in addition to their CP workload.
Despite the preference of the pharmaceutical companies for national submissions, they find the registration at the CP to be better and more efficient. Ease of communication has been reported for both the national and the CP although there is no direct access to the CP by the pharmaceutical companies. Most of the national regulatory authorities of the member states concur with the fact that the duration of the process to obtain market authorisation through the Centralised Procedure should be shorter to make it more appealing to applicants. The duration of the Centralised Procedure could be shortened by several means including increasing the number of CP committee meetings per year which has been clearly recognised by both the pharmaceutical companies and the national regulatory authorities. The lack of resources, mainly human expertise on the side of national regulatory authorities, contributes as well to the delays even though there is effective collaboration and sharing of information between the national regulatory authorities. Recruiting external experts can aid the national regulatory authorities with the shortage of human expertise, but there is a fear that this will impact the quality of the review. The demand for additional requirements by different national regulatory authorities adds to the delay and length of the process.
The most effective way to optimise the process of evaluation by the centralised authority would be by adopting an improved model. According to the regulatory authorities’ views, standardisation of the system would also be a means to improve and facilitate the regional registration process and maintain the supply of safe and effective medicines within a reasonable time frame. The national regulatory authorities in the Gulf have found the shared experience, ideas and knowledge at the centralised meetings to have enriched their own experience and knowledge which are reflected in their performance at the national level. The clear outcome of this collaboration and joint effort between the national regulatory authorities produces an improved scientific opinion and improved quality of decision-making. Pharmaceutical companies have also indicated their agreement with this where they found consistent evaluation of submissions and a better scientific opinion received from the centralised authority than from the national regulatory authorities leading to a more efficient system.
One major aspect of the CP approval reported by both the pharmaceutical companies and the member states is the centralised pharmacovigilance procedures and reporting system which are efficient and effective. In addition, member states of the centralised authority find the GCC guidelines to be sufficient and appropriate for their intended purpose. On the other hand, pharmaceutical companies, submitting to the centralised authority, struggle due to the absence of appropriate guidelines leading to inconsistency. Also there are differences when accessing the market at a national level such as re-requesting documents already submitted for the CP or not even approving the product for distribution after attaining GCC-DR registration.
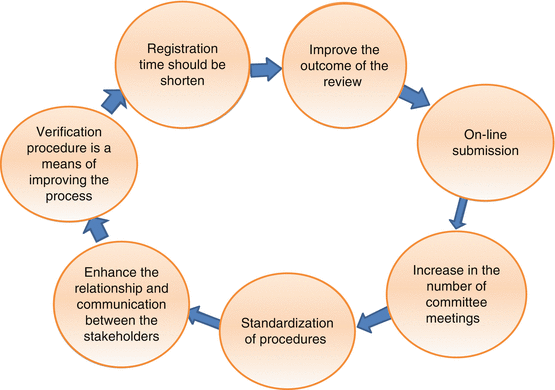
Certain aspects of the registration procedure can be modified in order to move forward and improve the quality of the experience. Both the national regulatory authorities of the member states and the pharmaceutical companies have agreed that the use of electronic online submissions at the centralised authority, as well as electronic communication between the member states, is one way to expedite the process. This is the current method implemented in the recognised established authorities worldwide such as the EMA and the FDA. Further, recognition of the assessment by established mature regulatory authorities through implementation of the verification procedure is a means of improving the process and easing the burden of the high workload on the national regulatory authorities as well as for the pharmaceutical companies. Harmonisation of requests and clarifications sent to the pharmaceutical companies is yet another way agreed by both the pharmaceutical companies and the national regulatory authorities. The national regulatory authorities identified the need for training, seminars and workshops to improve the experience of their expert staff. Pharmaceutical companies have suggested the same where the additional interactions between pharmaceutical companies and the national regulatory authorities would improve the overall knowledge of the national regulatory authorities especially as the pharmaceutical companies have worldwide experience. Also it would further enhance the relationship and communication between them. The pharmaceutical companies even suggested helping to draft, improve and refine guidelines including GMP. One of the key concerns, as highlighted by the companies, is the limited number of committee meetings per year. The quarterly meetings to validate the pharmaceutical products are clearly insufficient to meet the increasing demands for product registration and approval. The companies recommended that the frequency of the GCC-DR committee meetings needs to be 6 per year or every 45 days. In addition, subcommittees could be formed with more frequent meetings to handle specialised matters such the registration of life-saving products.
The companies recommended that the comments and opinions of the countries, where the products have been registered nationally, should be considered when products are being evaluated at GCC-DR. In this way, the approval process would be expedited, and this would facilitate faster approvals at a national level after the products have been approved by the GCC-DR. Over half of the companies supported the GCC-DR procedure, whilst 5 companies out of 30 preferred the national registration system. The main reason companies move towards local registration is because of the delay and queuing at the CP. Nevertheless, the companies agreed that if the GCC-CP improves, it will increase its acceptability of the marketing authorisation issued centrally in other markets. They also suggested the use of university laboratories to speed up ‘product testing’ against additional fees.
The only way to improve support for the GCC registration is to give some advantages over the national registration for companies who choose this route. It is very important to note that most companies are seeking to establish a single registration system, either national or centralised, for their products. Dealing with two registration systems in the same region is impractical. For all these reasons, companies usually adopt the most economically viable registration system that meets their corporate objectives. In conclusion, Fig. 10.1 summarises the key issues to be considered in the new improved GCC model.