to understand the ways that drugs work to affect biological systems, as a basis for safe and effective prescribing,
 to appreciate that pharmacology cannot be fully understood without a parallel understanding of related biological and clinical sciences, including biochemistry, physiology and pathology,
to appreciate that pharmacology cannot be fully understood without a parallel understanding of related biological and clinical sciences, including biochemistry, physiology and pathology, to develop numeracy skills for calculating drug doses and dilutions, and to enable accurate comparison of the relative benefits and risks of different drugs,
to develop numeracy skills for calculating drug doses and dilutions, and to enable accurate comparison of the relative benefits and risks of different drugs, to be able to comprehend and participate in research studies advancing knowledge of better treatment of patients.
to be able to comprehend and participate in research studies advancing knowledge of better treatment of patients.The answer to the frequently asked question ‘What do I need to know?’ will depend upon the individual requirements of the course you are studying, your year of study and the examinations you will be taking. The depth and type of knowledge required in different areas and topics may vary as you progress through your studies; for example, early in the course you might not be required to have detailed knowledge of drug monitoring, but you should know whether a drug has a narrow safety margin between its wanted and unwanted effects. Your personal enthusiasm for pharmacology is important and should be driven by the recognition that prescribing medicines is the commonest intervention that most doctors (and, increasingly, other health professionals) make to improve the health of their patients.
Learning about pharmacology is best approached using a variety of resources, in a range of learning scenarios and preferably in the context of clinical care, not from memorising lists of drug facts. We suggest that the following items cover the types of information that you should aim to encounter:


 the way the drug works (its mechanism of action and its clinical effects) and whether these vary significantly among patients,
the way the drug works (its mechanism of action and its clinical effects) and whether these vary significantly among patients,



 the absorption, distribution, metabolism and excretion (‘ADME’) of the drug (its pharmacokinetics), particularly where these show unusual characteristics,
the absorption, distribution, metabolism and excretion (‘ADME’) of the drug (its pharmacokinetics), particularly where these show unusual characteristics, the drug’s unwanted effects, including its propensity to cause interactions with other drugs or foods,
the drug’s unwanted effects, including its propensity to cause interactions with other drugs or foods,
The key reference for prescribers in the UK is the British National Formulary (BNF), available online at www.bnf.org/bnf/index.htm, which contains monographs for nearly all drugs licensed for use in the UK. The Appendix at the end of this chapter provides a formulary of core drugs in each drug class, which gives students in the early stages of training a manageable list of the drugs they are most likely to encounter in clinical practice.
Receptors and receptor-mediated mechanisms
Pharmacology is a materialist science in the sense that it describes how the material (physical) interaction of drug molecules with their macromolecular targets (‘receptors’) in the body modifies cellular processes to generate a desired effect. Drugs have been designed to interact with many different types of macromolecules, which evolved to facilitate endogenous signalling between cells, tissue and organs. The activities of most cellular processes are closely controlled to optimise homeostatic conditions in relation to physiological and metabolic requirements. Control can be divided typically into three main stages.
Each of these three stages provides important targets for drug action and this chapter will outline the principles underlying drug action mainly in stages 2 and 3.
Actions of drugs at binding sites (receptors)
For very many drugs the first step in producing a biological effect is by interaction of the drug with a receptor, either on the cell membrane or inside the cell, and it is this binding that triggers the cellular response. Drugs may be designed to mimic, modify or block the actions of endogenous ligands at that receptor. The receptor table at the end of this chapter shows that cell-membrane and cytosolic receptors tend to occur in different families (receptor types), reflecting their evolution from common ancestral receptors. Within any one family of receptors, different receptor subtypes have evolved divergently to facilitate increasingly specific signalling and distinct biological effects. As might be expected, different receptor families have different characteristics, but subtypes within each family retain common family traits.
In pharmacology, the perfect drug would be one that binds only to one type or subtype of receptor and consistently produces only the desired biological effect, without the unwanted effects that can occur when drugs bind ‘off target’. Although this ideal is impossible to attain, it has proved possible to develop drugs that bind avidly to their target receptor to produce their desired effect and have very much less (but not zero) ability to bind to other receptors, even ones within the same family, which might produce unwanted effects.
Where a drug binds to one type of receptor in preference to another it is said to show selectivity of binding or selectivity of drug action. Selectivity is never absolute but is high with some drugs and low with others. A drug with a high degree of selectivity is likely to show a greater difference between the dose required for its biological action and the dose that produces an unwanted or toxic action.
Major types of receptors
Despite the great structural diversity of drug molecules, most act on the following major types of receptors to bring about biological change.
 Transmembrane ion channels. These control the passage of ions across membranes and are widely distributed.
Transmembrane ion channels. These control the passage of ions across membranes and are widely distributed. Seven-transmembrane (heptahelical) receptors. This is a large family of receptors, most of which signal via guanine nucleotide-binding proteins (G-proteins). Following activation by a ligand, second messenger substances are formed which can bring about cellular molecular changes, including the opening of transmembrane ion channels.
Seven-transmembrane (heptahelical) receptors. This is a large family of receptors, most of which signal via guanine nucleotide-binding proteins (G-proteins). Following activation by a ligand, second messenger substances are formed which can bring about cellular molecular changes, including the opening of transmembrane ion channels. Enzyme-linked transmembrane receptors. This is a family of transmembrane receptors with an integral or associated enzymic component, such as a kinase or phosphatase. They signal changes in cells by phosphorylating or dephosphorylating intracellular proteins, thereby altering their activity.
Enzyme-linked transmembrane receptors. This is a family of transmembrane receptors with an integral or associated enzymic component, such as a kinase or phosphatase. They signal changes in cells by phosphorylating or dephosphorylating intracellular proteins, thereby altering their activity.
It should be noted that some mechanisms, such as the opening of ion channels, can be operated by direct interactions of drugs with the channel, or by G-protein-coupled mechanisms occurring as a first step with subsequent intracellular events activating the ion channels.
Transmembrane ion channels
Transmembrane ion channels that create pores across phospholipid membranes are ubiquitous and allow the transport of ions into and out of cells. The intracellular concentrations of ions are controlled by a combination of ion pumps and transporters, which transport specific ions from one side of the membrane to the other in an energy-dependent manner, and ion channels, which open to allow the selective, passive transfer of ions down their concentration gradients. Based on concentration gradients across the cell membrane:
 both Na+ and Ca2+ ions will diffuse into the cell if the channels are open, making the electrical potential of the cytosol more positive and causing depolarisation of excitable tissues,
both Na+ and Ca2+ ions will diffuse into the cell if the channels are open, making the electrical potential of the cytosol more positive and causing depolarisation of excitable tissues, K+ ions will diffuse out of the cell, making the electrical potential of the cytosol more negative and inhibiting depolarisation,
K+ ions will diffuse out of the cell, making the electrical potential of the cytosol more negative and inhibiting depolarisation,
The two major families of channel are the ligand-gated ion channels (LGICs) and the voltage-gated ion channels (VGICs; also called ionotropic receptors). LGICs are opened by the binding of a ligand, such as acetylcholine, to an extracellular part of the channel. VGICs are opened at particular membrane potentials by voltage-sensing segments of the channel. Both channel types can be targets for drug action. Both LGICs and VGICs can control the transport of a specific ion, but a single type of ion may be transported by more than one type of channel, including both LGIC and VGIC types. The complexity that has evolved can be seen in the example of the multiple types of K+ channel listed in Table 8.1.
LGICs include nicotinic acetylcholine receptors, γ-aminobutyric acid (GABA) receptors, glycine receptors and serotonin (5-hydroxytryptamine) 5-HT3 receptors. They are typically pentamers, with each subunit comprising four transmembrane helices clustering around a central channel or pore. Each peptide subunit is orientated so that hydrophilic chains face towards the channel and hydrophobic chains towards the membrane lipid bilayer. Binding of an agonist to the receptor causes a conformational change in the protein and results in extremely fast opening of the ion channel. The nicotinic acetylcholine receptor is a good example of this type of structure (Fig. 1.1). It requires the binding of two molecules of acetylcholine for channel opening. Channel opening lasts only milliseconds because the ligand rapidly dissociates and is inactivated. Drugs may modulate LGIC activity by binding directly to the channel, or indirectly by acting on G-protein-coupled receptors (see below) with the subsequent intracellular events then affecting the status of the LGIC.
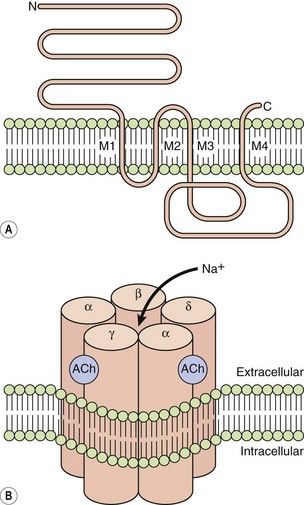
Fig. 1.1 The acetylcholine nicotinic receptor, a typical ligand-gated transmembrane ion channel.
(A) The receptor is constructed from subunits with four transmembrane regions (M1–M4). (B) Five subunits are assembled into the ion channel, which has two sites for acetylcholine binding, each formed by the extracellular domains of two adjacent subunits. On acetylcholine binding, the central pore undergoes conformational change that allows selective Na+ ion flow down its concentration gradient into the cell. N, amino terminus; C, carboxyl terminus.
VGICs include Ca2+, Na+ and K+ channels. The latter consist of four peptide subunits, each of which has between two and six transmembrane helices; in Ca2+ and Na+ channels there are four domains, each with six transmembrane helices, in a single large protein. The pore-forming regions of the transmembrane helices are largely responsible for the selectivity of the channel for a particular ion. Both Na+ and K+ channels are inactivated after opening; this is produced by an intracellular loop of the channel, which blocks the open channel from the intracellular end. The activity of VGICs may thus be modulated by drugs acting directly on the channel, such as local anaesthetics which maintain Na+ channels in the inactivated site by binding at the intracellular site (Ch. 18). Drugs may also modulate VGICs indirectly via intracellular signals from other receptors. For example, L-type Ca2+ channels are inactivated directly by calcium channel blockers, but also indirectly by drugs which reduce intracellular signalling from β1-adrenoceptors (see Fig. 5.5).
The ability of highly variable transmembrane subunits to assemble in a number of configurations leads to the existence of many different subtypes of channel for a single ion. For example, there are many different voltage-gated Ca2+ channels (L, N, P/Q, R and T types).
Seven-transmembrane receptors
Also known as 7TM receptors or the heptahelical receptor family, this is an extremely important group of receptors since the human genome has about 800 sequences for 7TM receptors and they are the targets of about 40% of modern drugs. The structure of a hypothetical 7TM receptor is shown in Figure 1.2; the N-terminal region of the polypeptide chain is on the extracellular side of the membrane and the polypeptide traverses the membrane seven times with helical regions, so that the C terminus is on the inside of the cell. The extracellular loops provide the receptor site for an appropriate agonist (a natural ligand or a drug), the binding of which alters the three-dimensional conformation of the receptor protein. The intracellular loops are involved in coupling this conformational change to the second messenger system, usually via a heterotrimeric G-protein, giving rise to the term G-protein-coupled receptor (GPCR).
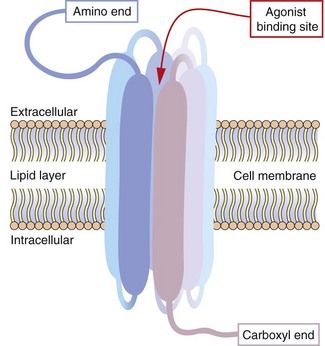
Fig. 1.2 Hypothetical seven-transmembrane (7TM) receptor.
The 7TM receptor is a single polypeptide chain with its amino (N-) terminus outside the cell membrane and its carboxyl (C-) terminus inside the cell. The chain is folded such that it crosses the membrane seven times, with each hydrophobic transmembrane region shown here as a thickened segment. The hydrophilic extracellular loops create a confined three-dimensional environment in which only the appropriate ligand can bind. Other potential ligands may be too large for the site or show much weaker binding characteristics. Selective ligand binding causes conformational change in the three-dimensional form of the receptor, which activates signalling proteins and enzymes associated with the intracellular loops, such as G-proteins and nucleotide cyclases.
The G-protein system: The heterotrimeric G-protein system (Fig. 1.3) consists of α, β and γ subunits.
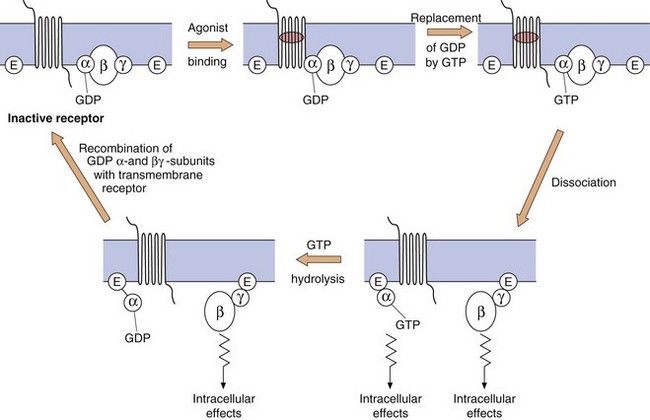
Fig. 1.3 The functioning of G-protein subunits.
Ligand (agonist) binding results in replacement of GDP on the α-subunit by GTP and the dissociation of the α- and βγ-subunits, each of which can affect a range of intracellular systems (shown as E on the figure) such as second messengers (e.g. adenylyl cyclase and phospholipase C), or other enzymes and ion channels (see Figs 1.4 and 1.5). Hydrolysis of GTP to GDP inactivates the α-subunit, which then recombines with the βγ-dimer to reform the inactive receptor.
 The α-subunit. More than 20 different types have been identified, belonging to four families (αs, αi, αq and α12/13). The α-subunit is important because it binds GDP and GTP in its inactive and active states, respectively; it also has GTPase activity, which is involved in terminating its own activity. When an agonist binds to the receptor, GDP (which is normally present on the α-subunit) is replaced by GTP. The active α-subunit–GTP dissociates from the βγ-subunits and can activate enzymes such as adenylyl cyclase. The α-subunit–GTP complex is inactivated when the GTP is hydrolysed back to GDP by the GTPase.
The α-subunit. More than 20 different types have been identified, belonging to four families (αs, αi, αq and α12/13). The α-subunit is important because it binds GDP and GTP in its inactive and active states, respectively; it also has GTPase activity, which is involved in terminating its own activity. When an agonist binds to the receptor, GDP (which is normally present on the α-subunit) is replaced by GTP. The active α-subunit–GTP dissociates from the βγ-subunits and can activate enzymes such as adenylyl cyclase. The α-subunit–GTP complex is inactivated when the GTP is hydrolysed back to GDP by the GTPase. The βγ-complex. There are many different isoforms of β- and γ-subunits that can combine into dimers, the normal function of which is to inhibit the α-subunit when the receptor is unoccupied. When the receptor is occupied by a ligand, the βγ-complex dissociates from the α-subunit and can itself activate cellular enzymes, such as phospholipase C. The α-subunit–GDP and βγ-subunit then recombine with the receptor protein to give the inactive form of the receptor–G-protein complex.
The βγ-complex. There are many different isoforms of β- and γ-subunits that can combine into dimers, the normal function of which is to inhibit the α-subunit when the receptor is unoccupied. When the receptor is occupied by a ligand, the βγ-complex dissociates from the α-subunit and can itself activate cellular enzymes, such as phospholipase C. The α-subunit–GDP and βγ-subunit then recombine with the receptor protein to give the inactive form of the receptor–G-protein complex.Second messenger systems: Second messengers are the key distributors of an external signal, as they are released into the cytosol and are responsible for affecting a wide variety of intracellular enzymes, ion channels and transporters. There are two complementary second messenger systems (Fig. 1.4).
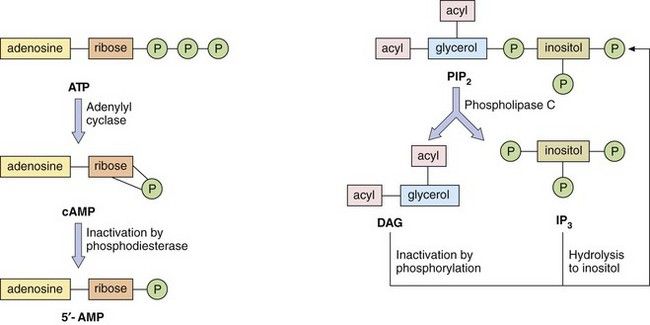
Fig. 1.4 Second messenger systems.
Stimulation of GPCRs produces intracellular changes by activating or inhibiting cascades of second messengers. Examples are cyclic adenosine monophosphate (cAMP), diacylglycerol (DAG) and inositol triphosphate (IP3) formed from phosphatidylinositol 4,5-bisphosphate (PIP2). See also Figure 1.5.
Cyclic nucleotide system: One system is based on cyclic nucleotides, such as:
 cyclic adenosine monophosphate (cAMP), which is synthesised from adenosine triphosphate (ATP) by adenylyl cyclase; cAMP induces numerous cellular responses by activating protein kinase A (PKA), which phosphorylates proteins, many of which are enzymes; phosphorylation can either activate or suppress cell activity;
cyclic adenosine monophosphate (cAMP), which is synthesised from adenosine triphosphate (ATP) by adenylyl cyclase; cAMP induces numerous cellular responses by activating protein kinase A (PKA), which phosphorylates proteins, many of which are enzymes; phosphorylation can either activate or suppress cell activity;
There are many isoforms of adenylyl cyclase; these show different tissue distributions and could be important sites of selective drug action in the future. The cyclic nucleotide second messenger (cAMP or cGMP) is inactivated by hydrolysis by phosphodiesterase (or PDE) isoenzymes to give AMP or GMP. There are 11 different families of phosphodiesterase isoenzymes, some of which are currently the targets of important drug groups (Table 1.1).
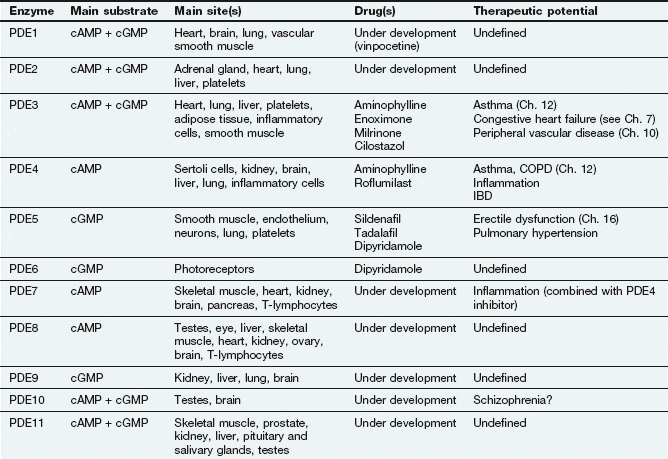
The phosphatidylinositol system: The other second messenger system is based on inositol 1,4,5-triphosphate (IP3) and diacylglycerol (DAG), which are synthesised from the membrane phospholipid phosphatidylinositol 4,5-bisphosphate (PIP2) by phospholipase C (Fig. 1.4). There are a number of isoenzymes of phospholipase C, which may be activated by the α-subunit–GTP or βγ-subunits of G-proteins. The main function of IP3 is to mobilise Ca2+ in cells. With the increase in Ca2+ brought about by IP3, DAG is able to activate protein kinase C (PKC) and phosphorylate target proteins. IP3 and DAG are then inactivated and converted back to PIP2.
Which second messenger systems are activated when a GPCR binds a selective ligand depends primarily on the nature of the Gα-subunit, as illustrated in Figure 1.5:
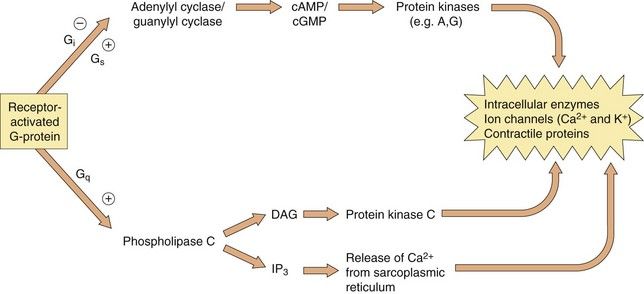
Fig. 1.5 The intracellular consequences of receptor activation.
The second messengers cAMP, cGMP, DAG and IP3 produce a number of intracellular changes, either directly, or indirectly via actions on protein kinases (which phosphorylate other proteins) or by actions on ion channels. The pathways can be activated or inhibited depending upon the type of receptor and G-protein and the particular ligand stimulating the receptor. The effect of the same second messenger can vary depending upon the biochemical functioning of cells in different tissues.

 Gi/o: inhibition of adenylyl cyclase (reduces cAMP), inhibition of Ca2+ channels, activation of K+ channels,
Gi/o: inhibition of adenylyl cyclase (reduces cAMP), inhibition of Ca2+ channels, activation of K+ channels,

The βγ-complex also has signalling activity: it can activate phospholipases and modulate some types of K+ and Ca2+ channels.
Activation of these second messenger systems by G-protein subunits thus affects many cellular processes such as enzyme activity (either directly or by altering gene transcription), contractile proteins, ion channels (affecting depolarisation of the cell) and cytokine production. The many different isoforms of Gα, Gβ and Gγ proteins may represent important future targets for selective drugs.
It is increasingly recognised that GPCRs may assemble into dimers of identical 7TM proteins (homodimers) or into heterodimers of different receptor proteins; the functional consequences of GPCR dimerisation and its implications for drug therapy are unclear.
Protease-activated receptors: Protease-activated receptors (PARs) are GPCRs stimulated unusually by a ‘tethered ligand’ located within the N terminus of the receptor itself, rather than by an independent ligand. Proteolysis of the N-terminal sequence by serine proteases such as thrombin, trypsin and tryptase enables the residual tethered ligand to bind to the receptor within the second extracellular loop (Fig. 1.6). To date, four protease-activated receptors (PAR 1–4) have been identified, each with distinct N-terminal cleavage sites and different tethered ligands. The receptors appear to play roles in platelet activation and clotting (Ch. 11), and in inflammation and tissue repair. Most of the actions of PAR are mediated by Gi, Gq and G12/13.
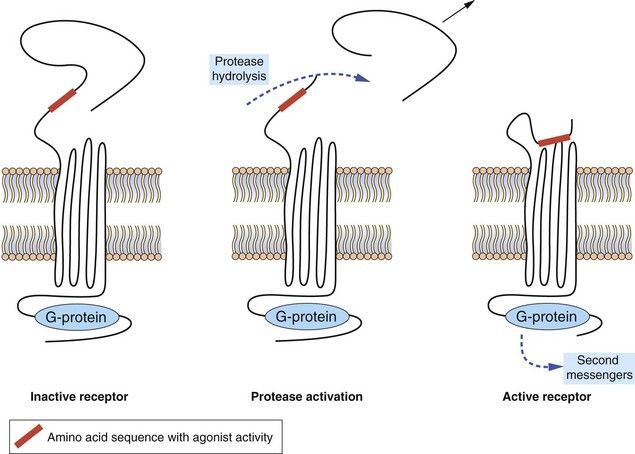
Fig. 1.6 Protease-activated receptors (PARs).
These GPCRs are activated by proteases such as thrombin which hydrolyse the extracellular peptide chain to expose a segment that acts as a tethered ligand (shown in red) and activates the receptor. The receptor is inactivated by phosphorylation of the intracellular (C-terminal) part of the receptor protein.
Enzyme-linked transmembrane receptors
Enzyme-linked receptors, most notably the receptor tyrosine kinases, are similar to the GPCRs in that they have a ligand-binding domain (or LBD) on the surface of the cell membrane, they traverse the membrane and they have an intracellular effector region (Fig. 1.7). They differ from GPCRs in their extracellular ligand-binding site, which is very large to accommodate their polypeptide ligands (including hormones, growth factors and cytokines), and in having only one transmembrane helical region. Importantly, their intracellular action requires a linked enzymic domain, most commonly an integral kinase domain which activates the receptor itself or other proteins by phosphorylation. Activation of enzyme-linked receptors enables binding and activation of many intracellular signalling proteins, leading to changes in gene transcription and in many cellular functions. There are five families of enzyme-linked transmembrane receptors.
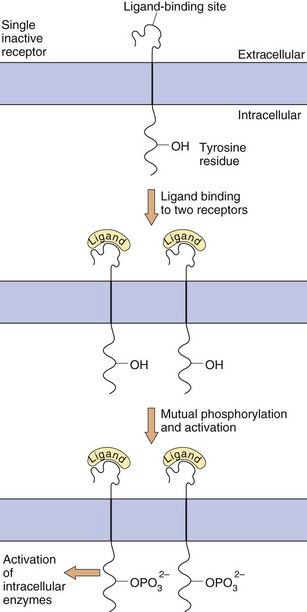
Fig. 1.7 Enzyme-linked transmembrane receptors.
This receptor tyrosine kinase has a large extracellular domain, a single transmembrane segment and an integral kinase domain. Ligand binding causes phosphorylation of tyrosine residues on the receptor and on other target proteins, leading to intracellular changes in cell behaviour. Other enzyme-linked receptors have tyrosine phosphatase, serine-threonine kinase or guanylyl cyclase enzymic activity.
 Receptor tyrosine kinase (RTK) family: ligand binding causes receptor dimerisation and transphosphorylation of tyrosine residues within the receptor itself and sometimes in associated cytoplasmic proteins. Up to 20 classes of RTK include receptors for growth factors, many of which signal via proteins of the mitogen-activated protein (MAP) kinase cascade, leading to effects on gene transcription, apoptosis and cell division. Constitutive over-activity of an RTK called Bcr-Abl causes leucocyte proliferation in chronic myeloid leukaemia, which is treated with imatinib, a drug that blocks the uncontrolled RTK activity.
Receptor tyrosine kinase (RTK) family: ligand binding causes receptor dimerisation and transphosphorylation of tyrosine residues within the receptor itself and sometimes in associated cytoplasmic proteins. Up to 20 classes of RTK include receptors for growth factors, many of which signal via proteins of the mitogen-activated protein (MAP) kinase cascade, leading to effects on gene transcription, apoptosis and cell division. Constitutive over-activity of an RTK called Bcr-Abl causes leucocyte proliferation in chronic myeloid leukaemia, which is treated with imatinib, a drug that blocks the uncontrolled RTK activity. Tyrosine phosphatase receptor family: they dephosphorylate tyrosines on other transmembrane receptors or cytoplasmic proteins; they are particularly common in immune cells.
Tyrosine phosphatase receptor family: they dephosphorylate tyrosines on other transmembrane receptors or cytoplasmic proteins; they are particularly common in immune cells. Tyrosine kinase-associated receptor family (or non-receptor tyrosine kinases): these lack integral kinase activity but activate separate kinases associated with the receptor; examples include inflammatory cytokine receptors and signalling via the Jak/Stat pathways to affect inflammatory gene expression.
Tyrosine kinase-associated receptor family (or non-receptor tyrosine kinases): these lack integral kinase activity but activate separate kinases associated with the receptor; examples include inflammatory cytokine receptors and signalling via the Jak/Stat pathways to affect inflammatory gene expression. Receptor serine-threonine kinase family: activation of these phosphorylates serine and threonine residues in target cytosolic proteins; everolimus is a serine-threonine kinase inhibitor used in renal and pancreatic cancer.
Receptor serine-threonine kinase family: activation of these phosphorylates serine and threonine residues in target cytosolic proteins; everolimus is a serine-threonine kinase inhibitor used in renal and pancreatic cancer.
Intracellular (nuclear) receptors
Many hormones act at intracellular receptors to produce long-term changes in cellular activity by altering the genetic expression of enzymes, cytokines or receptor proteins. Such hormones are lipophilic to facilitate their movement across the cell membrane. Examples include the thyroid hormones and the large group of steroid hormones, including glucocorticoids, mineralocorticoids and the sex steroid hormones. Their actions on DNA transcription are mediated by interactions with intracellular receptors (Table 1.2) located either in the cytoplasm (type 1) or the nucleus (type 2).
Table 1.2
Some families of intracellular receptors
| Type 1 (cytoplasmic) | |
| Oestrogen receptors | ER (α, β) |
| Progesterone receptors | PR (A, B) |
| Androgen receptor | AR |
| Glucocorticoid receptor | GR |
| Mineralocorticoid receptor | MR |
| Type 2 (nuclear) | |
| Thyroid hormone receptors | TR (α, β) |
| Vitamin D receptor | VDR |
| Retinoic acid receptors | RAR (α, β, γ) |
| Retinoid X receptors | RXR (α, β, γ) |
| Liver X (oxysterol) receptors | LXR (α, β) |
| Peroxisome proliferator-activated receptors | PPAR (α, γ, δ) |
The intracellular receptor typically includes a highly conserved DNA-binding region with zinc-containing loops and a variable LBD (Table 1.3). The sequence of hormone binding and action for type 1 intracellular receptors is shown in Figure 1.8. Type 1 receptors are typically found in an inactive form in the cytoplasm linked to chaperone proteins such as heat-shock proteins (HSPs). Binding of the hormone induces conformational change in the receptor; this causes dissociation of the HSP and reveals a nuclear localisation sequence (or NLS) which enables the hormone–receptor complex to pass through nuclear membrane pores into the nucleus. Via their DNA-binding domain, the active hormone–receptor complexes can interact with hormone response elements (HRE) at numerous sites in the genome. Binding to the HRE usually activates gene transcription, but sometimes it silences gene expression and decreases mRNA synthesis.
Table 1.3
The structure of steroid hormone receptor proteins
| Section of protein | Domain | Role |
| A/B | N-terminal regulatory domain | Regulates transcriptional activity |
| C | DNA-binding domain | Highly conserved; binds receptor to DNA by two zinc-containing regions |
| D | Hinge region | Enables intracellular translocation |
| E | Ligand-binding domain | Enables specific ligand binding; also binds chaperone proteins and facilitates receptor dimerisation |
| F | C-terminal domain | Highly variable; unknown function |
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


