Primary Oxalosis
Shane M. Meehan, MBBCh
Key Facts
Etiology/Pathogenesis
Autosomal recessive mutations of AGT (PO1) or GR (PO2) genes
Clinical Issues
PO1: Hyperoxaluria (> 100 mg/d), increased urinary glycolate, renal failure
PO2: Hyperoxaluria, increased urinary L-glyceric acid, mild renal failure
Diagnosis by assay of enzyme activity in liver tissue
End-stage renal failure frequent in PO1
Macroscopic Features
Stones are composed of > 95% calcium oxalate monohydrate (whewellite)
Microscopic Pathology
Extensive birefringent crystal deposits, in rosettes and sheaves, in tubules and interstitium with giant cell reaction and fibrosis
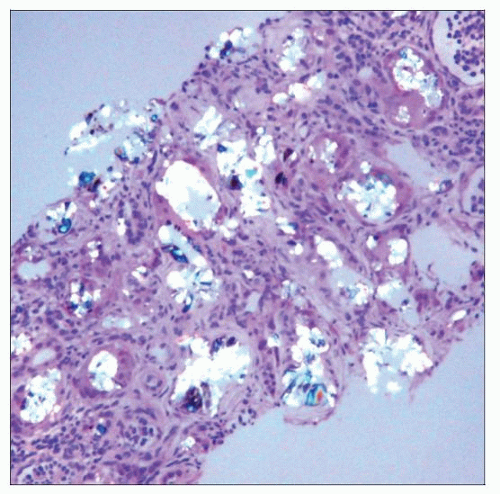 Hematoxylin & eosin using partially polarized light shows abundant anisotropic calcium oxalate in the cortex of a 4-month-old male with primary oxalosis type 1. |
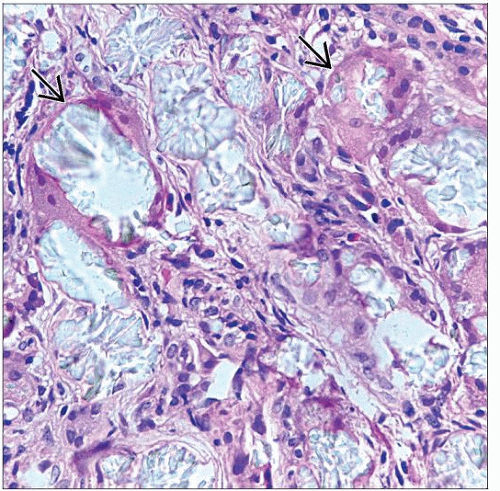 Refractile, pale yellow, calcium oxalate crystals are seen, some of which are in giant cells  in the renal cortex. in the renal cortex. |
TERMINOLOGY
Abbreviations
Primary oxalosis (PO)
Type 1 (PO1), type 2 (PO2)
Synonyms
Primary hyperoxaluria (PH), primary hyperoxalemia
Definitions
Oxalate overproduction due to gene mutations affecting enzymes that catalyze glyoxylate breakdown, with systemic calcium oxalate deposition
ETIOLOGY/PATHOGENESIS
Hereditary Disorder
Autosomal recessive mutations of alanine glyoxylate aminotransferase gene (PO1) or glyoxalate reductase gene (PO2)
Alanine glyoxylate aminotransferase (AGT) gene on chromosome 2q36-37
AGT converts glyoxylate to glycine in hepatic peroxisomes
Glyoxylate reductase (GR) gene on chromosome 9c
GR converts glyoxylate to glycolate in hepatocyte cytoplasm
Mutations are multiple (50 identified for AGT and 14 for GR), have varying effects
e.g., loss of catalytic function, accelerated degradation, and mistargeting
Loss of function of these enzymes results in excess glyoxylate, which is converted to oxalate by lactate dehydrogenase
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


