Fig. 20.1
Metabolites of heroin and half-lives in plasma that need to be considered in the design of heroin vaccines
Fig. 20.2
Clinically used opiates that are related to heroin. (a) Structural analogs of heroin. (b) Pharmaceutical medications used in the treatment of heroin addiction and overdose
We have attached the linker to positions C3 and C6 and the bridge nitrogen of heroin/morphine surrogates in our efforts to develop a heroin vaccine (Fig. 20.3) (Matyas et al. 2013, 2014; Li et al. 2014). For attachment at the C3 position, 6-PrOxyHap was selected and synthesized with a thiol-amide linker at the C3 position (Fig. 20.3a). It contains a 2-oxopropyl group at C6 as a stable mimic of 6AM (Li et al. 2014). Immunization with 6-PrOxyHap conjugated to tetanus toxoid (TT) produced high titer antibody responses that cross-reacted with 6AM, heroin, morphine, and codeine. 6-PrOxyHap presented the C6 and bridge nitrogen portion of opiate structure as the front face to the immune system, while the immune system was unable to recognize the C3 linker attachment point or back face. Consequently, the induced antibodies have a subsite that binds to the 2-oxopropyl group that can also accommodate the acetyl group of 6AM and heroin and the hydroxyl group of morphine and codeine. Since there is no recognition of the C3 by the antibodies induced by 6-PrOxyHap, the C3 methyl ether of codeine and the acetyl of heroin do not inhibit binding. C3 linkers were also tested by Spector et al. and Koida et al., who observed cross-reactivity with codeine and morphine (Spector and Parker 1970; Koida et al. 1974a). Although 6-PrOxyHap induced high titer antibodies with the expected cross-reactivity to other opiates, the ability of heroin to effectively inhibit pain in antinociception assays of 6-PrOxyHap immunized mice was only partially blunted (Li et al. 2014). This suggests that C3 may not be the optimal linkage site for a vaccine against heroin or that the 2-oxopropyl group at C6 is not an adequate substitute of the C6 acetyl of heroin and 6AM.
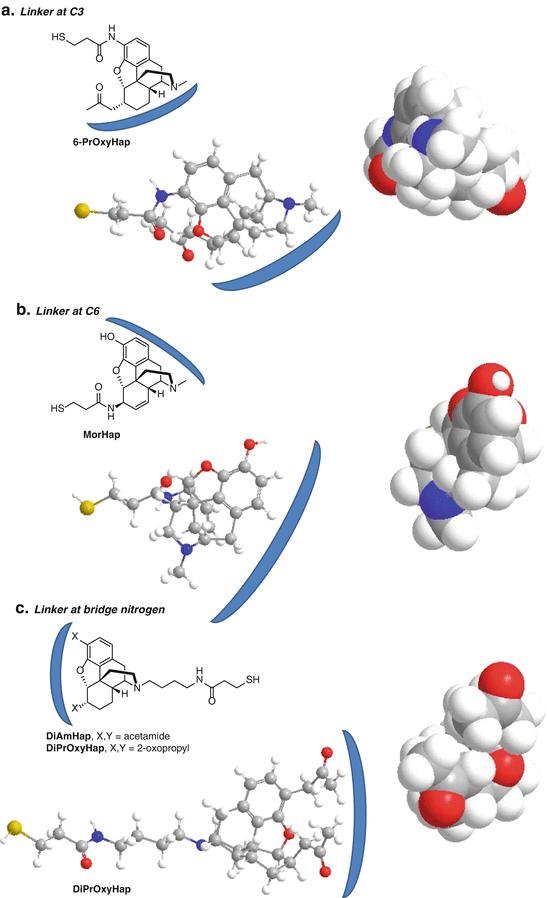
Fig. 20.3
Relevant hapten–protein conjugates that we have used as potential heroin vaccines. The blue arcs denote the front face. The space-filling models were created in ChemBio3D Ultra with a minimized energy calculation. They are oriented with the linker pointing back into the page, and the front face is forward depicting the portion seen by the antibody (coming straight down). The ball-and-stick models are rotated 90° counterclockwise from the space-filling models (a) Linker at C3. (b) Linker at C6. (c) Linker at bridge nitrogen
Numerous investigators have used C6-linked haptens (Wainer et al. 1973; Koida et al. 1974b; Bonese et al. 1974; Akbarzadeh et al. 1999, 2009; Farhangi et al. 2012; Anton and Leff 2006; Li et al. 2011). We developed a hapten called MorHap that contains a thiol-amide linker at the C6 of morphine (Fig. 20.3b) (Matyas et al. 2013, 2014). Mice immunized with MorHap-TT conjugates exhibited high titer antibodies and reduced antinociceptive effects caused by injection of heroin. Using the strategies described in this chapter, we have effectively abolished the antinociceptive effects due to heroin challenge (Jalah et al., 2015). Competitive ELISA showed MorHap-induced antibodies that cross-reacted with 6AM and morphine but very poorly cross-reacted with heroin (Matyas et al. 2014). The antibodies also did not cross-react with codeine (Matyas et al., unpublished). Since MorHap is linked at the C6 position (back face), the C3 and bridge nitrogen (front face) are presented to the immune system. The cross-reactivity of the induced antibodies with 6AM and morphine is undoubtedly due to the binding to the C3 hydroxyl and the bridge nitrogen. Heroin contains a C3 acetyl group and codeine a C3 methyl ether which cannot be accommodated in the C3 hydroxyl subsite of the MorHap-induced antibodies.
Bridge nitrogen-linked haptens of morphine also have been investigated and induced antibodies that were specific for morphine (Morris et al. 1975; Findlay et al. 1981; Usagawa et al. 1993). In an effort to synthesize a stable hapten that would induce antibodies that cross-react with heroin, we synthesized two bridge nitrogen-linked haptens, DiPrOxyHap and DiAmHap (Fig. 20.3c) (Matyas et al. 2014; Li et al. 2014). DiPrOxyHap contains a 2-oxopropyl group at both the C3 and C6 positions, whereas DiAmHap contains an amido group at both the C3 and C6 positions. Attachment of the linker to the bridge nitrogen presented the C3 and C6 groups (front face) to the immune system, whereas the bridge nitrogen (back face) was not presented. Immunization with DiPrOxyHap or DiAmHap conjugated to TT induced very high antibody titers in mice and partially reduced the antinociceptive effects from heroin challenge. Antibodies induced by DiPrOxyHap cross-reacted with heroin and 6AM, but not with morphine or codeine (Li et al. 2014). However, the cross-reactivities with heroin and 6AM were only observed at higher concentrations in competitive ELISA suggesting that the binding to heroin and 6AM was lower affinity. Heroin, 6AM, or morphine did not inhibit the binding of DiAmHap-induced antibodies to DiAmHap-BSA-coated ELISA plates (Matyas et al. 2014). Subsequent competitive ELISA of DiAmHap-induced antibodies demonstrated that heroin, 6AM, and morphine inhibited the binding of the antibodies to MorHap-BSA-coated ELISA plates. This suggests that the binding affinity of the DiAmHap-induced antibodies to DiAmHap is very high and that the cross-reactivity to heroin, 6AM and morphine, although present, is low affinity.
We have demonstrated that by using the concept of facial recognition, it is possible to understand the antibody specificities obtained following immunization with stable heroin/morphine haptens containing linkers at different positions. Based on these observations, it may be possible to use the concept of facial recognition to design a hapten that would be expected to induce antibodies of a desired specificity. This concept is not only applicable to the design of a heroin vaccine but may also be useful for the design of other substance abuse vaccines or other hapten-based vaccines.
20.2 Hapten–Protein Conjugates
Congruent with hapten design, the carrier protein must be selected at the onset of the development process because the immunological potency of a given hapten–protein conjugate may not be translatable to other carriers. What are the “practical” characteristics of an ideal carrier protein? The ideal carrier protein for vaccines against drugs of abuse (1) must be safe for human use, (2) should generate reproducible hapten–protein conjugates of consistent hapten density, (3) can be characterized using established analytical methods, and (4) should have acceptable yield of the hapten–protein conjugate. Since assessment of vaccine efficacy utilizes animals or human volunteers which give variable responses, immunization without quantitative analysis of the hapten–protein conjugates can lead to unpredictable and greatly irregular responses. This section will address the intricate relationships of hapten design and carrier proteins and how these relationships determine the reproducibility and yield of the hapten–protein conjugates.
20.2.1 Carrier Proteins
What are the attributes of an ideal carrier for a conjugate vaccine, particularly a substance abuse vaccine?
1.
The carrier should be a homogenous, nonaggregated protein that can be characterized by analytical techniques.
2.
It should be readily able to be produced under current Good Manufacturing Practices to allow for human use.
3.
It should be reproduced by recombinant molecular biology methods or isolated from nonvertebrate sources to eliminate concerns of adventitious agents.
4.
It should be safe for human use, inducing no adverse reactions following immunization and, if possible, should be beneficial to the recipient.
5.
It should be highly immunogenic to all recipients without genetic restrictions and should induce potent immune responses following the initial immunization.
6.
It should not induce cross-reactive immune responses to normal recipient molecules, including proteins, lipids, carbohydrates, DNA, etc.
7.
Preexisting immunity should not negatively impact the immune response to the conjugate.
8.
A previous record of clinical use would be very beneficial to ensure the above criteria.
The five carriers that have been used for licensed polysaccharide conjugate vaccines are diphtheria toxoid (DT), a genetically modified cross-reacting material of diphtheria toxin (CRM197), tetanus toxoid (TT), meningococcal outer membrane complex (OMPC), and Hemophilus influenza protein D (Knuf et al. 2011; Pichichero 2013; Tontini et al. 2013). These carriers have been extensively studied and are safe for human use, including babies and children. These polysaccharide conjugate vaccines are very different than substance abuse vaccines, since polysaccharide conjugate vaccines contain large oligosaccharides conjugated to the carrier protein and are not single small molecule hapten-based vaccines. Nevertheless, the safety profiles of the carrier protein and its ability to induce potent T-cell-dependent antibody response should be readily applicable to substance abuse vaccines. OMPC induced high levels of antibody following primary immunization, which further increased after the second dose, but failed to further increase with further boosts (Einhorn et al. 1986; Weinberg et al. 1987). Since substance abuse vaccines are expected to need continuing boosts to maintain antibody levels, OMPC may not be suitable as a carrier. Both TT and DT are formaldehyde treated to detoxify the tetanus toxin and diphtheria toxin, respectively (Knuf et al. 2011; Pichichero 2013). In addition to being carriers in conjugate vaccines, they are the antigens in the licensed tetanus and diphtheria vaccines, respectively, which are potent and safe (Barkin et al. 1985). CRM197 is a detoxified mutant of DT (Malito et al. 2012). Although there are a few exceptions, preexisting immunity to TT, DT, or CRM197 does not appear to have a significant negative impact on the potency or efficacy of the polysaccharide vaccines (Pichichero 2013; Pollabauer et al. 2009; Borrow et al. 2011; Pobre et al. 2014). These carrier proteins meet all of the above-listed criteria for substance vaccine carriers. TT, DT, and CRM197 have been used as carriers for preclinical studies for substance abuse vaccines and have generally induce potent immune responses (Matyas et al. 2013, 2014; Li et al. 2014; Anton and Leff 2006; Moreno et al. 2010; McCluskie et al. 2013; Pravetoni et al. 2014) establishing them as acceptable protein carriers.
Other carriers, such as bovine serum albumin (BSA), keyhole limpet hemocyanin (KLH), recombinant cholera toxin B, disrupted adenovirus serotype 5 (Ad5) proteins or hexons, and recombinant Pseudomonas aeruginosa exoprotein A (rEPA), have been used as carriers for substance abuse vaccines (Alving et al. 2014). Among these carriers, BSA has been used extensively in preclinical studies involving drugs of abuse. BSA, a 66 kDa protein, is an attractive carrier for vaccine development because it (1) contains 30–35 surface lysines amenable for conjugation (Hermanson 2008), (2) has high aqueous solubility, (3) is commercially available in acceptable purity, and (4) is relatively cheap. In 2009 and 2012, a team of Iranian scientists, who used 6SM-BSA to vaccinate morphine addicts, reported successful clinical trials (Akbarzadeh et al. 2009; Farhangi et al. 2012). However, BSA is not suitable for human vaccines in the United States or Europe due to the risk of prions and other adventitious agents associated with bovine products.
KLH has been extensively studied in human clinical trials as a carrier for therapeutic cancer vaccines (Curigliano et al. 2006; Kantele et al. 2011). It is isolated from the hemolymph of the inedible mollusk, Megathura crenulata, also known as giant keyhole limpets. It is a cylindrical, copper-containing molecule responsible for oxygen transportation (Swaminathan et al. 2014). KLH is a mixture of two isoforms, both of which are didecamers, consisting of 400 kDa glycoproteins (Swerdlow et al. 1996; Gatsogiannis and Markl 2009). It contains ~2000 lysines allowing for easy conjugation of haptens (Hermanson 2008). However, it has a tendency to precipitate and aggregate. Due to the large molecular weight of the particle, it is very difficult to quantify the haptens conjugated to the carrier. The tendency of KLH to precipitate and aggregate makes it difficult to handle and characterize in a vialed vaccine. Recently, monomers or dimers of KLH have been produced using recombinant technology and may eliminate some of the disadvantages of the high molecular weight KLH, but the recombinant KLH may be less immunogenic (Lebrec et al. 2014). Nevertheless, the use of KLH as a carrier for preclinical studies of they vaccines has successfully demonstrated the principal that substance vaccines can have efficacy against substances of abuse. Its use as a carrier protein on the clinical path for licensure remains unclear due to the source, its tendency to aggregate, and the inability to determine the number of haptens attached.
The B subunit of the cholera toxin has been tested as a carrier for a candidate cocaine vaccine in human clinical trials through a phase III efficacy trial, where it failed to have efficacy in cocaine addicts (Martell et al. 2009; Haney et al. 2010; Kosten et al. 2002, 2014a). Cholera toxin is produced by Vibrio cholera (Spangler 1992). It is the causative agent of cholera in which the secreted toxin binds to the intestinal epithelial cells resulting in the production of cyclic AMP and the subsequent excessive secretion of fluid. The B subunit of cholera toxin is a pentamer of 11 kDa protein. Each monomer binds to the ganglioside GM1. The pentamer binding results in a very high, nearly irreversible, binding to the cell surface, and it has been reported to have modest adjuvant activity, binding to both dendritic cells and lymphocytes and causing activation (Isomura et al. 2005; Luci et al. 2006; Hou et al. 2014). The binding site for GM1 contains lysine as a critical residue for GM1 recognition (Merritt et al. 1994). It is important that these lysines are not used to conjugate the substance abuse hapten, but it is difficult to control the conjugation to lysines during the conjugation process. This may represent one of the reasons why the cocaine vaccine was not efficacious. The cholera toxin B subunit has been used in numerous clinical trials with a good safety record (Martell et al. 2009; Haney et al. 2010; Kosten et al. 2002, 2014a; Sun et al. 2010) and represents a potential carrier protein for substance abuse vaccines if lysine conjugation is not used for attachment of the hapten.
Disrupted and partially purified adenovirus serotype 5 (Ad5) proteins or capsids have been used as carriers for experimental cocaine and nicotine vaccines (Koob et al. 2011; Hicks et al. 2011; Wee et al. 2012; Maoz et al. 2013; Rosenberg et al. 2013; De et al. 2013; Hicks et al. 2014). They elicited potent antibody titers, which block the effects of cocaine and nicotine. There are a number of concerns related to the use of Ad5 as a carrier. First, the partially purified proteins and capsids are mixture of proteins, which makes it very difficult to reproducibly manufacture the hapten conjugated vaccine and very difficult to quantify the number of haptens per carrier molecule. In addition, the use of carbodiimide coupling chemistry would be expected to lead to cross-linking of the proteins in the formulation, further making it difficult to quantify the number of conjugated haptens. The rationale of selecting a pulmonary virus as a carrier for vaccines of drugs of abuse that are typically taken by the respiratory route is intriguing, allowing for the development of a mucosal antibody response which will potentially block the drug from entering the bloodstream. However, it is noted that in 2008, an HIV vaccine trial that used Ad5 as vector was halted due to futility (Buchbinder et al. 2008; McElrath et al. 2008; Robb 2008). The HIV vaccine did not prevent HIV infection and had an increased incidence of HIV infection rates in male participants who received vaccine compared to placebo. Analysis indicated that preexisting immunity to Ad5 was one of the factors that was associated with increased HIV infection rates (Buchbinder et al. 2008; Cheng et al. 2012). Further analysis indicated that preexisting Ad5 immunity significantly reduced the immune response to Ad5 vaccines (Cheng et al. 2012). The net effect of these studies is that Ad5 has been replaced by other Ad vectors, such as Ad26, which do not naturally infect humans.
Pseudomonas aeruginosa exoprotein A (rEPA) contains a single amino acid deletion of glutamic acid at position 553 of exotoxin A (Lukac et al. 1988). Similar to CRM197, the deletion effectively blocks the toxic ribosyltransferase activity. It is a 67 kDa protein that has been used in clinical trials as a carrier for polysaccharide vaccines (Passwell et al. 2010; Thiem et al. 2011; Rondini et al. 2011; Szu et al. 2014), but to date, there is no licensed vaccine that uses rEPA as a carrier. However, it has also been tested in clinical trials as a carrier for a nicotine vaccine (NicVAX®) by Nabi Biopharmaceuticals (Hatsukami et al. 2011; Hoogsteder et al. 2012, 2014). Vaccination with NicVAX® gave promising results through phase II trials, but in 2011, Nabi Biopharmaceuticals announced that the phase III trials failed to achieve its primary endpoint and that there was no statistical difference between the treated and placebo groups (NicVAX® phase III trial result. 2015). Conjugate vaccines based on rEPA are well tolerated with generally good safety profiles in humans. However, given the failure of NicVAX® and the lack of licensure of any other conjugate vaccine utilizing rEPA, the utility of rEPA as a carrier for substance abuse vaccines remains questionable.
What is the preferred carrier for substance abuse vaccines? Based on the criteria listed at the beginning of this section, the preferred carrier proteins would be TT and CRM197. Both have been extensively used in licensed polysaccharide-based conjugate vaccines and have an excellent safety profile. The downside to TT is that it is formaldehyde treated, which reduces the number of surface lysines available for conjugation. However, this allows for increased spacing among the attached haptens on the surface. We believe that the best strategy is to test multiple carriers at different hapten densities to the select the best carrier for a given hapten.
20.2.2 Conjugation Chemistry
Azo coupling (Matsushita et al. 1974; Matsukura et al. 1975; Castro et al. 1980, 1985a, b), photoactivatable cross-linking (Ettinger et al. 1997), carbodiimide-mediated conjugation, imine formation (Beike et al. 1999), thioalkylation using halogenated compounds (Ramakrishnan et al. 2014), and maleimide–thiol chemistry (Matyas et al. 2014; Stowe et al. 2011b; Pravetoni et al. 2012a; Moreno et al. 2011; Carroll et al. 2011) have been used for the preparation of hapten–protein conjugates. Azo coupling involves in situ generation of hapten-diazonium species, which react rapidly with tyrosine, histidine, and lysine residues of proteins (Tabachnick and Sobotka 1959), and the aminophenyl hapten precursor itself. The fast kinetics of “self-conjugation” may result in poor reproducibility of the coupling reactions and the harsh conditions (alkaline pH, 0 °C) used for coupling may also denature the carrier.
In the photoactivatable cross-linking approach, aryl azide is attached to the surface lysines of the carrier protein and generates nitrenes when exposed to UV light (250–350 nm). Nitrenes are highly reactive and can react in multiple ways with the hapten or the proteins, such as addition to double bonds, insertion into C-H and N-H, and ring expansion followed by nucleophilic attack (Gilchrist and Charles 1969; Sinz 2006). Because nitrene reactions give a multitude of possible products, the hapten–protein conjugates may not be precisely reproduced. Exposure of the carrier to UV may also damage key epitopes. In addition, this approach may be difficult to scale up for the production of a vaccine.
Carbodiimide chemistry, which involves the formation of an amide bond between an activated acid and amine (Hoare and Koshland 1966), is a popular conjugation strategy in vaccines against drugs of abuse. In this strategy, a carboxylic acid moiety is synthesized into the haptenic surrogate and reacted with surface lysines. Since the carboxylic acid itself is present in proteins (i.e., aspartic and glutamic acids), intermolecular amide bond formation that results in insoluble oligomers can also take place. In addition, carbodiimide chemistry is not compatible for some haptens possessing a primary or secondary amine, as in the case of amphetamine.
Imine formation uses a homobifunctional linker that possesses carbonyl groups at both ends (DeCaprio et al. 1987). The surface lysines of the protein are allowed to react with the linker, and the resulting intermediate is conjugated with an amino hapten. Like carbodiimide-mediated conjugation, imine formation is prone to intramolecular oligomerization. Thioalkylation using halogenated compounds involves cysteine and a halogenated hapten as the reacting partners. However, this strategy is not frequently used because thiols in proteins are predominantly in an oxidized state and cysteine residues are less abundant in proteins (Stephanopoulos and Francis 2011). The maleimide–thiol chemistry (Smyth et al. 1964) employs a heterobifunctional linker with N-hydroxysuccinimide (NHS) and maleimide at its ends for coupling to surface lysines and thiols, respectively. In contrast to carbodiimide, a thiol is attached on the framework of the molecule to obtain the surrogate hapten. As the maleimide–thiol chemistry does not produce oligomerization (Hermanson 2008), the yield of the protein conjugates could be solely assessed in terms of hydrophobicity of the hapten–protein conjugates. For these reasons, we believe that the maleimide–thiol chemistry is the optimal coupling strategy. In addition, Fontaine et al. have recently reported a method to stabilize maleimide–thiol conjugates in vivo through ring opening of the succinimide moiety (Fontaine et al. 2015).
20.2.3 Hapten Density and Conjugate Yield
Hapten density, defined as the number of haptens covalently attached at the surface of a carrier molecule, is pivotal to vaccine efficacy. The effect of hapten density on the immunological properties of small molecule hapten–protein conjugates was elegantly demonstrated from the works of Klaus et al. (Klaus and Cross 1974a, b; Klaus and Mitchell 1974). Klaus et al. showed that dinitrophenyl (DNP)-BSA conjugates with hapten densities of less than 30 induced a modest primary response, an IgM to IgG switch, and concomitant hapten-specific memory, while highly substituted conjugates with hapten density of 50 elicited a dose-dependent primary IgM response, little IgG antibody, and little or no memory. The immunogenicity of small molecule–protein conjugates of dopamine (DA), 2,4-dichlorophenoxyacetic acid (2,4-D), and atrazine (AT) was also reported to be dependent on the hapten density. Malaitsev et al. reported that DA-BSA conjugates with hapten density of 6–14 produced the optimal immune response (Malaitsev and Azhipa 1993). On the other hand, DA-BSA with low hapten density (4) and higher hapten density (18–22) yielded lower antibody titers. Boro et al. showed that a significantly higher titer was obtained from 2,4-D-BSA conjugates with a hapten density of 20 relative to higher density (31) and lower density (2–9) conjugates of 2,4-D-BSA conjugates (Boro et al. 2009). Rajesh et al. demonstrated that atrazine–protein conjugates with hapten density of 15–30 evoke higher antibody titer with moderate antibody affinities (Rajesh et al. 2013). Adamczyk et al. argued that a lower hapten density conjugate induced a slower immune response but often led to an antibody with a higher affinity, while a high hapten density conjugate resulted in an IgM response which exceeded that of IgG and produced antibodies of lower affinity (Adamczyk et al. 1996). Pryde et al. and Carroll et al. also reported the effect of hapten density on the efficacy of vaccines against drugs of abuse. Pryde et al. showed that hapten density is correlated to the functional efficacy of anti-nicotine vaccines (Pryde et al. 2013). In general, nicotine–DT conjugates with a hapten density of <9 induced either low-titer or low-affinity antibodies, while hapten densities of 13–19 generated antibodies that retained nicotine in the plasma. Carroll et al. demonstrated that active immunization of METH-BSA conjugates with hapten density of 5 and 12 hapten densities generated IgG responses in 10 % and 90 % of the mice respondents, respectively (Carroll et al. 2011). The findings of Carroll et al. infer that responses would be less variable if optimal hapten density conjugates were used. Overall, these studies indicated the importance of hapten density on vaccine efficacy and demonstrate that the optimal density must be determined for each given hapten and carrier.
Using maleimide–thiol chemistry, we were able to synthesize hapten–protein conjugates with reproducible hapten densities (Torres et al. 2014). Recently, we demonstrated that hapten density (1) can be controlled using appropriate conjugation protocols and stoichiometry, (2) is a determinant of protein yield for MorHap-BSA conjugates, and (3) is crucial to ELISA absorbance as the plate coating antigen. We further investigated if the optimized procedure is applicable to TT. Different hapten density profiles were observed for BSA and TT conjugates (Fig. 20.4). These results could be explained by the difference in size between BSA and TT. By trinitrobenzenesulfonic acid (TNBS) assay, the surface lysines of TT (~27–35) and BSA (~30–35) are comparable. The size of TT (152 kDa) is more than twice the size of BSA (66 kDa), and consequently, the “effective concentration” of surface amines would be relatively lower in TT. The protein yield profile for MorHap-BSA and MorHap-TT conjugates showed that hapten density determines the yield of the reaction (Fig. 20.5). BSA with a MorHap density of 34 exhibited the lowest yield, while TT and BSA conjugates with 22–24 MorHap attached gave quantitative yields.
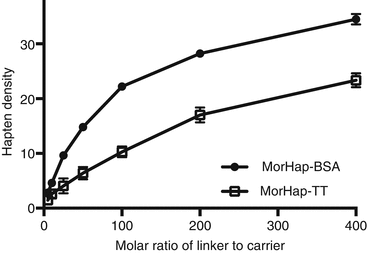
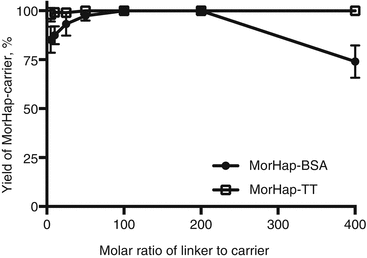
Fig. 20.4
Effect of linker ratios on hapten density of MorHap conjugates. BSA and TT were treated with varying molar ratios of the SM-(PEG)2. Excess linker was removed by spin desalting column. The maleimide-BSA and maleimide-TT were reacted with 100-fold excess of MorHap. Excess hapten was removed by dialysis. Hapten density was determined using MALDI-TOF MS
Fig. 20.5
Effect of linker ratios on protein yield of MorHap conjugates. BSA and TT were treated with varying molar ratios of the SM-(PEG)2. Excess linker was removed by spin desalting column. The maleimide-BSA and maleimide-TT were reacted with 100-fold excess of MorHap. Excess hapten was removed by dialysis. Protein yield was measured by BCA
Is the optimized procedure applicable to other haptens? We further tested our optimized procedure by coupling a more hydrophobic hapten, 6-PrOxyHap, to BSA, TT, and KLH. 6-PrOxyHap is a suitable model hapten to assess protein yield because most haptenic surrogates used for vaccines against drugs of abuse have high hydrophobicity. As anticipated the hapten profiles between BSA and TT were different (Fig. 20.6). The determination of hapten density of KLH conjugates was a formidable task as we were not able to accurately measure the molecular weight of 6-PrOxyHap-KLH conjugates by MALDI-TOF MS. Several groups have also reported the same challenge (Stowe et al. 2011b; Pravetoni et al. 2012b). Comparable hapten densities were observed for MorHap-BSA and 6-PrOxyHap-BSA as well as for MorHap-TT and 6-PrOxyHap-TT conjugates using the same coupling ratios. The yields of 6-PrOxyHap conjugates were significantly reduced relative to MorHap conjugates (Fig. 20.7). For example, 6-PrOxyHap-BSA and MorHap-BSA with ~34 haptens have 5 % and 74 % yields, respectively. Although 6-PrOxyHap-KLH conjugates have a higher yield as compared to 6-PrOxyHap-BSA and 6-PrOxyHap-TT conjugates, it is difficult to assess their conjugation advantage without the information on hapten density. Whether the high yield is due to lower hapten density or the ability of hapten–KLH conjugate to maintain high aqueous solubility is difficult to determine. Based on these findings, one can infer that KLH and BSA conjugates have different hapten densities even though they were treated with the same [linker]/[protein] molar ratios. Collectively, these results suggest that the stoichiometry of the reacting partners and the nature of the carrier protein mediate hapten density. Furthermore, protein yield is highly dependent on hapten density and hydrophobicity of the hapten.
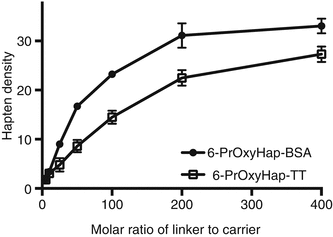
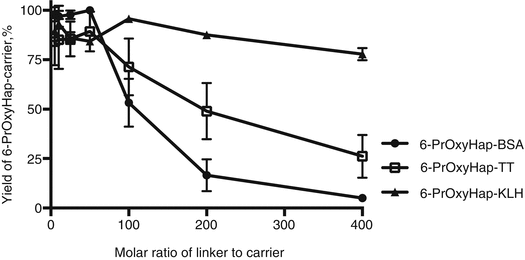
Fig. 20.6
Effect of linker ratios on hapten density of 6-PrOxyHap conjugates. BSA, TT and KLH were treated with varying molar ratios of the SM-(PEG)2. Excess linker was removed by spin desalting column. The maleimide-BSA, maleimide-TT, and maleimide-KLH were reacted with 100-fold excess of 6-PrOxyHap. Excess hapten was removed by dialysis. Hapten density was determined using MALDI-TOF MS. The hapten density of 6-PrOxyHap-KLH could not be accurately assessed by MALDI-TOF MS and is consequently not shown
Fig. 20.7
Effect of linker ratios on protein yield of 6-PrOxyHap conjugates. BSA, TT, and KLH were treated with varying molar ratios of the SM-(PEG)2. Excess linker was removed by spin desalting column. The maleimide-BSA, maleimide-TT, and maleimide-KLH were reacted with 100-fold excess of 6-PrOxyHap. Excess hapten was removed by dialysis. Protein yield was measured by BCA
20.2.4 Linker
The linker moiety in the hapten–linker–carrier complex can affect the immunological as well as the practical aspects of a vaccine formulation. The linker directly influences the presentation of the hapten to the immune system. A long linker can generate antibodies which have a deep binding site for the hapten and the target drug (Lim et al. 1998; Larsen et al. 2001; Celikel et al. 2009) as compared to a short linker which generates antibodies that bind the hapten near the surface of the antibody binding site (Pozharski et al. 2005). The linker can also impact the protein yield of the conjugate due to the hydrophobicity of the linker.
The nature and length of the linker must be considered during the design of the hapten–protein conjugate because the linker itself can serve as an additional antigenic determinant. Bridge effect or the co-recognition of the linker during antibody generation has been reported (Franek 1987). For heroin vaccines, aliphatic (Wainer et al. 1973; Koida et al. 1974b; Bonese et al. 1974; Akbarzadeh et al. 1999, 2009; Farhangi et al. 2012; Anton and Leff 2006; Li et al. 2011; Morris et al. 1975; Findlay et al. 1981; Usagawa et al. 1993; Beike et al. 1999), aromatic (Spector et al. 1973; Gross et al. 1974), and polyethylene glycol (PEG)-based (Matyas et al. 2013, 2014; Li et al. 2014) linkers have been used. We do not recommend the use of aromatic linker as they may induce antibodies with promiscuous binding sites for aromatic ligands (James et al. 2003). Although aliphatic linkers do not contain functional groups, Gorris et al. showed that long, aliphatic chains can be recognized by antibodies (Gorris et al. 2011). In fact, Gorris et al. have exploited the bridge effect to enhance the affinity of the antibodies to 2,4-dinitrophenol (2,4-DNP) and demonstrated that the affinity of the antibodies for 2,4-DNP increased with increasing linker length. We were not able to obtain hapten–TT conjugates with a high hapten density (i.e., 30–35) using aliphatic linkers due to the poor aqueous solubility of the resulting hapten–TT conjugates.
PEG-based molecules are attractive linkers for conjugating haptens to carriers because: (1) They have low immunogenicity (Schellekens et al. 2013) and PEG-based materials have been used as drug delivery agents (Gombotz and Pettit 1995). (2) They are hydrophilic molecules and thereby increase the solubility of the hapten–protein conjugate. The synthesis of conjugates with high hapten density, even when the hapten itself is hydrophobic, is feasible. There have been reports also that hydrophilic linkers better display the hapten to the immune system than hydrophobic linkers (Nara et al. 2008; Mercader et al. 2008). In addition, PEG linkers have been shown to improve hapten presentation at bilayer interfaces (Ahlers et al. 1992; Leckband et al. 1995; Jung et al. 2008). (3) SM (PEG)n series linkers with varying chain length are commercially available (Information on SM(PEG)n crosslinking reagents. 2015). Since NHS and maleimide moieties are already present in the linker, the synthesis, providing reproducible hapten–carrier conjugates using maleimide–thiol chemistry, becomes facile and economical (Torres et al. 2014).
What is the optimal nature and length of the linker for the drug hapten–protein conjugates? To address this problem, Pryde et al. synthesized nicotine hapten, H7, appended to 12 linkers of varying lipophilicity, hydrophilicity, and flexibility (Pryde et al. 2013). Surprisingly, none of the 12 linkers gave a better functional response to the hapten. It is difficult to dissect the “linker effect,” if there is any, because H7-DT conjugates of different hapten densities were compared. As discussed vide supra, hapten density affects the immunogenicity of conjugates. A systematic approach to assess “linker effect” would be a rigorous head-to-head comparison of hapten–protein conjugates with similar hapten densities. In our opinion, there is an optimal PEG linker length that would generate a superior immune response. However, this should be determined empirically once the lead hapten design has been identified.
20.2.5 Choosing the Lead Hapten–Protein Conjugate
The commercial feasibility of a vaccine formulation depends heavily on the (1) hapten design, (2) carrier protein, and (3) resulting hapten–protein conjugate. (1) The hapten design must strike a balance among immunogenicity, stability, and yield of hapten–carrier conjugate. Inadequate presentation of the hapten will lead to low-titer and low-affinity antibodies and unwarranted cross-reactivity. Labile haptens will compromise the reproducibility and potency of the vaccine. Highly hydrophobic haptens will result in low yields of the hapten–protein conjugates that would make them difficult to characterize. (2) Safety, cost, and the ability to induce potent antibody responses are the major concerns for choosing the carrier. Proven immunologically potent protein carriers that are already present in FDA licensed vaccines should be selected to avoid unanticipated safety issues during clinical trials. (3) The resulting hapten–protein conjugates should utilize simple conjugation schemes that result in acceptable yield, and the conjugates should be easily characterized using routine analytical methods, such as MALDI-TOF MS, chemical assays, and liquid chromatography. Finally, hapten density should be measured as one of the “quality control tests” for vaccine release to ensure batch-to-batch consistency of the vaccine formulation.
20.3 Adjuvants
Substance abuse vaccines require that the vaccine induce antibodies of both high titer and high affinity and that the antibodies must be of long duration. In general, both the haptens and the protein carriers, by themselves, are poorly immunogenic. Consequently, an adjuvant is required to stimulate the immune response to the hapten, and the proper selection of the adjuvant is critical to the development of the vaccine to substances of abuse (Alving et al. 2014). While aluminum salts have been widely utilized worldwide as an adjuvant in numerous licensed vaccines against a variety of diseases (Brito et al. 2013; Brito and O’Hagan 2014; Oleszycka and Lavelle 2014; Powell et al. 2015), the limited experience with human responses to aluminum salt-adjuvanted vaccines for drugs of abuse suggests that the immune responses may be too low in magnitude and duration for the development of a successful vaccine (Alving et al. 2014; Martell et al. 2009; Hatsukami et al. 2011; Cornuz et al. 2008; Kosten et al. 2014b). An adjuvant or combination of adjuvants that is safe and potent for enhancing both the magnitude and durability of the immune response is therefore needed. It also should be easily available, easily manufactured, and cost-effective. Although many potential experimental adjuvants have been proposed, our approach has been to utilize generic adjuvant formulations that would mimic or enhance the compositions, safety, and efficacy properties of adjuvants present in licensed vaccines. In our experience, the adjuvants that are present in licensed proprietary vaccines generally can be reproduced in generic forms with considerable freedom to operate regarding intellectual property.
Adjuvants in licensed vaccines: As described by Alving et al. (Alving et al. 2014), there are only six types of adjuvants in licensed vaccines:
Aluminum salts are the most commonly used adjuvants (Brito et al. 2013; Brito and O’Hagan 2014; Oleszycka and Lavelle 2014; Powell et al. 2015; Baylor et al. 2002).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


