Potassium
There is an important inter-relationship between the processes of potassium metabolism and those of water and sodium balance, renal function and acid-base disorders (discussed in the preceding chapters). Abnormalities of potassium homeostasis such as plasma concentrations that are too low (hypokalaemia) or too high (hyperkalaemia) are also relatively common in clinical practice and important to know about, as they may become life threatening.
POTASSIUM HOMEOSTASIS
The total amount of potassium in the body is about 3000 mmol, of which about 98 per cent is intracellular. For this reason the plasma potassium concentration is a poor indicator of the total body content.
Factors affecting plasma potassium concentration
The intracellular potassium ion concentration ([K+]) is large and provides a reservoir for the extracellular compartment. Consequently changes in water balance have little direct effect on the plasma [K+], unlike plasma [Na+].
The normal potassium intake is about 60-100 mmol/day. Potassium enters and leaves the extracellular compartment by three main routes:
the intestine,
the kidneys,
the membranes of all other cells.
The intestine
Potassium is principally absorbed in the small intestine. Dietary intake replaces net urinary and faecal loss. Prolonged starvation can cause or aggravate potassium depletion leading to hypokalaemia. Meat, vegetables and fruit including bananas have about 6.2 mmol/100 g of potassium. Foods with a high content of potassium (more than 12.5 mmol/100 g) include dried fruits (dates and prunes), nuts, avocados and bran/wheat grain. Dried figs, molasses and seaweed are rich in potassium (more than 25 mmol/100 g).
Potassium leaves the extracellular compartment in all intestinal secretions, usually at concentrations near to or a little above that in plasma. A total of about 60 mmol/day is lost into the intestinal lumen, most of which is reabsorbed. Less than 10 mmol/day is present in formed faeces. Excessive intestinal potassium loss can occur in diarrhoea, ileostomy fluid or through fistulae.
The kidneys
Glomerular filtrate
Potassium is filtered by the glomeruli and, owing to the huge volume of the filtrate, about 800 mmol (about a quarter of the total body content) would be lost daily if there were no tubular regulation. The net loss is about 10 per cent of that filtered.
The tubules
Potassium is normally almost completely reabsorbed in the proximal tubules. Damage to these may cause potassium depletion. Potassium is secreted in the distal tubules and collecting ducts in exchange for Na+; hydrogen ions (H+) compete with potassium ions (K+). Aldosterone stimulates both exchange mechanisms. If the proximal tubules are functioning normally, potassium loss in the urine depends on three factors:
The amount of sodium available for exchange: this depends on the glomerular filtration rate, filtered sodium load, and sodium reabsorption from the proximal tubules and loops of Henle. As discussed later, reabsorption in the loops is inhibited by many diuretics.
The circulating aldosterone concentration: this is increased following fluid loss, with volume contrac tion, which usually accompanies intestinal loss of potassium, and in most conditions for which patients are receiving diuretic therapy. Hyperkalaemia stimulates aldosterone release in synergy with angiotensin II, while hypokalaemia inhibits it.
The cell membranes
The Na+/K+ adenosine triphosphatase (ATPase) ‘pump’ on cell surfaces maintains a high intracellular [K+]. This exchanges three Na+ ions from cells in exchange for two K+ ions in the extracellular fluid (ECF), thus establishing an electrochemical gradient across the cell membrane, with a net positive charge in the ECF. The loss of K+ from cells down the concentration gradient is opposed by this electrochemical gradient. Potassium is also exchanged for H+ (Fig. 5.1).
A small shift of K+ out of cells may cause a significant rise in plasma concentrations, whether in vivo or in vitro. In the latter situation, the artefactual hyperkalaemia due to haemolysed or old specimens may be misinterpreted and should be avoided. Usually the shift of K+ across cell membranes is accompanied by a shift of Na+ in the opposite direction, but the percentage change in extracellular [Na+] is much less than that of K+.
Increased uptake and net gain of K+ by cells may occur in alkalosis due to increased uptake by cells and increased urinary loss. Insulin enhances the cellular uptake of glucose and potassium. Hyperkalaemia stimulates insulin secretion and hypokalaemia inhibits it. This effect may be used to treat hyperkalaemia by giving exogenous insulin/glucose infusion (see Treatment of hyperkalaemia). It is the main cause of the change from hyperkalaemia to hypokalaemia during the treatment of diabetic ketoacidosis.
Catecholamines have a similar action, and it has been suggested that this may contribute to the hypokalaemia sometimes found after the stress of myocardial infarction. β-adrenergic stimulation increases cellular potassium uptake by stimulating the Na+/K+-ATPase ‘pump’. β-blockade increases plasma potassium concentration and β-agonists decrease it, an effect that is independent of body potassium stores.
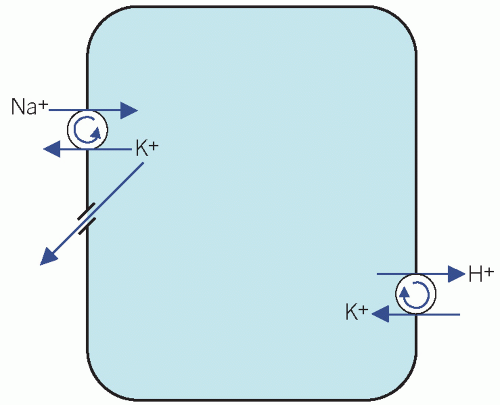 Figure 5.1 Potassium pumps on cell membranes. |
Synthesis of Na+/K+-ATPase is stimulated by thyroxine, which may contribute to the hypokalaemia sometimes associated with hyperthyroidism.
Relationship between hydrogen and potassium ions (Fig. 5.2)
The extracellular [H+] affects the entry of potassium into all cells. Changes in the relative proportions of K+ and H+ in distal renal tubular cells affect the urinary loss of potassium. There is a reciprocal relationship between K+ and H+:
In acidosis, increased loss of potassium from cells into the ECF coupled with reduced urinary secretion of potassium causes hyperkalaemia.
In alkalosis, net increased uptake of potassium into cells and increased urinary potassium loss cause hypokalaemia.
In the kidney, Na+ derived from the luminal fluid is pumped through the cell in exchange for either K+ or H+. If K+ is lost from the ECF, it passes down the increased concentration gradient, so reducing its
intracellular content. Unless there is renal tubular cell damage, Na+ are reabsorbed in exchange for fewer K+ and more H+ than usual. For each H+ formed within the tubular cell by the CD mechanism, one bicarbonate ion (HCO3−) is produced:
intracellular content. Unless there is renal tubular cell damage, Na+ are reabsorbed in exchange for fewer K+ and more H+ than usual. For each H+ formed within the tubular cell by the CD mechanism, one bicarbonate ion (HCO3−) is produced:
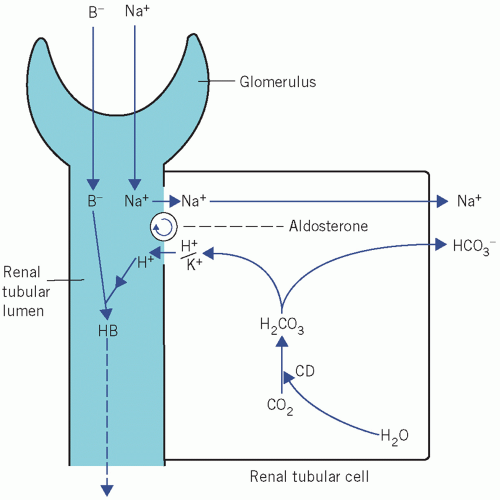 Figure 5.2 Exchange of Na+ for either K+ or H+ in the renal tubules. B−, non-bicarbonate base; CD, carbonate dehydratase. |
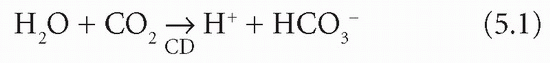
As more H+ is secreted into the urine, the reaction is increased and more HCO3− is generated, passing into the ECF accompanied by the reabsorbed Na+. The result is an extracellular alkalosis and acid urine. Therefore, chronic potassium depletion is usually accompanied by a high plasma bicarbonate concentration. The combination of hypokalaemia and a high plasma bicarbonate concentration is more likely to be due to K+ depletion than to metabolic alkalosis.
Potassium and diuretic therapy
Diuretics may be used to treat hypertension as well as oedema, for example in cardiac failure. They can be divided into two principal groups, based upon their site of action.
Potassium-losing diuretics increase the sodium load on the distal tubules and collecting ducts with enhanced Na+/K+ exchange. This may result in hypokalaemia.
Loop diuretics, such as furosemide or bumetanide, inhibit sodium reabsorption from the ascending limb of the loops of Henle.
Thiazides act at the junction of the loops and the distal tubules, which are sometimes called the ‘cortical diluting segments’.
Carbonate dehydratase inhibitors, such as acetazolamide, are rarely used as diuretics but are still used to treat glaucoma. They may cause a hypokalaemic hyperchloraemic acidosis.
Potassium-sparing diuretics.
Diuretics that inhibit either aldosterone directly or the exchange mechanisms in the distal tubules and collecting ducts cause potassium retention and may lead to hyperkalaemia, especially if glomerular function is impaired. Potassium-sparing/-retaining diuretics include spironolactone, a competitive aldosterone antagonist.
Other potassium-sparing diuretics include amiloride and triamterene, which are direct inhibitors of the Na+/K+ exchange mechanism in the distal tubules.
Potassium-sparing diuretics can be used together with those causing potassium loss when hypokalaemia cannot be controlled by potassium supplementation. Used alone, they have only a weak diuretic action. Beware of giving potassium supplements to patients taking potassium-sparing diuretics or angiotensin-converting enzyme (ACE) inhibitors or angiotensin II receptor blockers (ARBs) because of the risk of hyperkalaemia.
Measurement of plasma and urinary potassium
In rapidly changing clinical states frequent estimation of plasma potassium is the best way of assessing therapy. Plasma potassium is best measured in a freshly collected, unhaemolysed venous sample as a corresponding serum concentration is higher owing to the release of intracellular potassium as part of the clotting process. In chronic potassium depletion, measurement of the plasma bicarbonate concentration ([HCO3−]) may help to indicate the state of cellular repletion (intracellular potassium depletion causes extracellular alkalosis).
Urinary potassium (kaluria) estimations to determine the primary cause of potassium depletion may be useful diagnostically to help find the cause of hypokalaemia. Most extrarenal causes of potassium loss are associated with volume depletion and therefore with secondary hyperaldosteronism. Excretion of less than about 20 mmol/day can be expected only in the well-hydrated hypokalaemic patient with extrarenal losses, in whom aldosterone secretion is inhibited. Therefore a low potassium excretion confirms extrarenal loss, but a high one does not prove that it is the primary cause. High urinary potassium concentration (for example more than 20 mmol/L) in the face of hypokalaemia suggests inappropriate K+ renal loss as a cause of the hypokalaemia, for example due to renal tubular problems or mineralocorticoid excess syndromes.
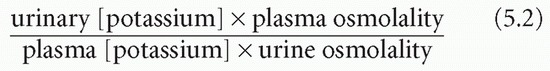
The transtubular potassium gradient (TTKG) is an estimate of the potassium concentration at the end of the cortical collecting duct beyond the site where aldosterone influences potassium secretion. This can be calculated on a spot urine (which should have an osmolality greater than that of the plasma) and plasma samples from the following equation:
ABNORMALITIES OF PLASMA POTASSIUM
Hypokalaemia
Hypokalaemia is often the result of potassium depletion, but if the rate of loss of potassium from cells equals or exceeds that from the ECF, potassium depletion may exist without hypokalaemia. Hypokalaemia can also occur without depletion if there is a shift of K+ into cells. The causes of hypokalaemia (summarized in Box 5.1) may be classified according to the predominant cause as either renal or non-renal. Pseudohypokalaemia is defined as the in vitro uptake of K+ from the plasma into blood cells, primarily erythrocytes. This sometimes occurs if the blood sample is kept in a warm environment. This contrasts with the more usual hyperkalaemia found in specimens stored in the refrigerator.
Renal causes of hypokalaemia
Enhanced renal secretion of potassium due to increased activity of the pump in distal renal tubules by mineralocorticoid activity. This is associated with a hypokalaemic alkalosis.
Primary hyperaldosteronism (Conn’s syndrome) due to adrenal adenoma or hyperplasia (see Chapter 8).
Secondary hyperaldosteronism often aggravates other causes of potassium depletion (see Chapter 2).
Cushing’s syndrome and steroid therapy. Patients secreting excess of, or on prolonged therapy with, glucocorticoids tend to become hypokalaemic due to the mineralocorticoid effect on the distal renal tubules (see Chapter 8).
‘Ectopic’ adrenocorticotrophic hormone (ACTH) secretion, which stimulates cortisol secretion (see Chapter 8).
Bartter’s syndrome is a rare autosomal recessive condition in which there is hyperplasia of the renal juxtaglomerular apparatus with increased renin and therefore aldosterone secretion. Angiotensin II activity is impaired and therefore there is no vasoconstriction; consequently the patient is normotensive. This latter finding helps to distinguish the syndrome from that of primary hyperaldosteronism, in which there can also be a hypokalaemic alkalosis. Individuals may have polyuria, polydipsia, short stature, learning difficulties and maternal polyhydramnios. The defect is in the epithelial ion channels of the ascending limb. The biochemical features mimic those of high-dose loop diuretic intake. Treatment consists of potassium supplementation, often with a potassium-sparing diuretic such as spironolactone or amiloride. By suppressing prostaglandin-stimulated renin release, non-steroidal anti-inflammatory drugs may also improve the condition.
The very rare Gitelman’s syndrome, in contrast to Bartter’s syndrome, is often asymptomatic until adulthood. It is an autosomal recessive condition due to a mutation in the thiazide-sensitive Na+/Cl− co-transporter in the distal convoluted tubules. Unlike Bartter’s, there is more severe hypomagnesaemia and hypocalciuria. Once thiazide use has been excluded, a hypokalaemic hypomagnesaemic alkalosis in the presence of a urinary calcium to creatinine molar ratio less than 0.2 is suggestive of Gitelman’s syndrome.
Liddle’s syndrome, also very rare, is an autosomal dominant condition with hypertension and hypokalaemic metabolic alkalosisis, in which suppressed plasma aldosterone and renin concentrations are associated with cerebrovascular disease. Mutations in the selective sodium channels cause increased sodium reabsorption through the collecting duct epithelial sodium channel, causing hypertension. Treatment involves sodium restriction and potassium-sparing diuretics such as amiloride or triamterene; spironolactone is ineffective.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


