Portal Hypertension and Cirrhosis
KEY CONCEPTS
![]() Cirrhosis is a severe, chronic, irreversible disease associated with significant morbidity and mortality. However, the progression of cirrhosis secondary to alcohol abuse can be interrupted by abstinence. It is therefore imperative for the clinician to educate and support abstinence from alcohol as part of the overall treatment strategy of the underlying liver disease.
Cirrhosis is a severe, chronic, irreversible disease associated with significant morbidity and mortality. However, the progression of cirrhosis secondary to alcohol abuse can be interrupted by abstinence. It is therefore imperative for the clinician to educate and support abstinence from alcohol as part of the overall treatment strategy of the underlying liver disease.
![]() Patients with cirrhosis should receive endoscopic screening for varices, and certain patients with varices should receive primary prophylaxis with nonselective β-adrenergic blockade therapy to prevent variceal hemorrhage.
Patients with cirrhosis should receive endoscopic screening for varices, and certain patients with varices should receive primary prophylaxis with nonselective β-adrenergic blockade therapy to prevent variceal hemorrhage.
![]() When nonselective β-adrenergic blocker therapy is used to prevent rebleeding, β-blocker therapy can be titrated to achieve a goal heart rate of 55 to 60 beats/min or the maximal tolerated dose.
When nonselective β-adrenergic blocker therapy is used to prevent rebleeding, β-blocker therapy can be titrated to achieve a goal heart rate of 55 to 60 beats/min or the maximal tolerated dose.
![]() Octreotide is the preferred vasoactive agent for the medical management of variceal bleeding. Endoscopy employing endoscopic band ligation is the primary therapeutic tool for the management of acute variceal bleeding.
Octreotide is the preferred vasoactive agent for the medical management of variceal bleeding. Endoscopy employing endoscopic band ligation is the primary therapeutic tool for the management of acute variceal bleeding.
![]() The combination of spironolactone and furosemide is the recommended initial diuretic therapy for patients with ascites.
The combination of spironolactone and furosemide is the recommended initial diuretic therapy for patients with ascites.
![]() All patients who have survived an episode of spontaneous bacterial peritonitis should receive long-term antibiotic prophylaxis.
All patients who have survived an episode of spontaneous bacterial peritonitis should receive long-term antibiotic prophylaxis.
![]() The mainstay of therapy of hepatic encephalopathy involves therapy to lower blood ammonia concentrations and includes diet therapy, lactulose, and antibiotics alone or in combination with lactulose.
The mainstay of therapy of hepatic encephalopathy involves therapy to lower blood ammonia concentrations and includes diet therapy, lactulose, and antibiotics alone or in combination with lactulose.
Chronic liver injury causes damage to normal liver tissue resulting in the development of regenerative nodules surrounded by fibrous bands.1 Cirrhosis is an advanced stage of liver fibrosis. The advanced fibrosis of cirrhosis leads to shunting of the portal and arterial blood supply directly into hepatic outflow through the central veins, and exchange between hepatic sinusoids and hepatocytes is compromised. Clinical consequences of cirrhosis include impaired hepatocyte function, the increased intrahepatic resistance of portal hypertension, and hepatocellular carcinoma. Circulatory irregularities such as splanchnic vasodilation, vasoconstriction and hypoperfusion of the kidneys, water and salt retention, and increased cardiac output also occur. The word cirrhosis is derived from the Greek kirrhos, meaning orange-yellow, and refers to the color of the cirrhotic liver as seen on autopsy or during surgery.2
![]() While cirrhosis has many causes (Table 24-1), in the Western world, excessive alcohol intake and hepatitis C are the most common causes.1,3 This chapter elucidates the pathophysiology of cirrhosis and the resultant effects on human anatomy and physiology. Treatment strategies for managing the most commonly encountered clinical complications of cirrhosis are discussed.
While cirrhosis has many causes (Table 24-1), in the Western world, excessive alcohol intake and hepatitis C are the most common causes.1,3 This chapter elucidates the pathophysiology of cirrhosis and the resultant effects on human anatomy and physiology. Treatment strategies for managing the most commonly encountered clinical complications of cirrhosis are discussed.
TABLE 24-1 Etiology of Cirrhosis

EPIDEMIOLOGY
The exact prevalence of cirrhosis is unknown, but a reasonable estimate is that 1% of populations have histologically diagnosable cirrhosis.1 Cirrhosis was responsible for over 31,000 deaths in America in 2010, and chronic liver disease continues to be ranked 12th among the leading causes of death in the United States.4 Acute variceal bleeding and spontaneous bacterial peritonitis (SBP) are among the immediately life-threatening complications of cirrhosis. Associated conditions causing significant morbidity include ascites and hepatic encephalopathy (HE). Approximately 50% of patients with cirrhosis develop ascites during 10 years of observation and, within 2 years, nearly half of patients who develop ascites will die.5
PATHOPHYSIOLOGY OF CIRRHOSIS
Any discussion of cirrhosis must be based on a firm understanding of hepatic anatomy and vascular supply. Conceptually, the liver can be thought of as an elaborate blood filtration system receiving blood from the hepatic artery and the portal vein (Fig. 24-1), with portal blood originating from the small intestines.6 Blood enters the liver via the portal triad, which contains branches of the portal vein, hepatic artery, and bile ducts. It then drains through the sinusoidal spaces (also known as the space of Disse) of the hepatic lobule (Fig. 24-2), which are lined by the workhorses of the liver, the hepatocytes. Individual hepatocytes are arranged in plates that are one cell thick and organized around individual central veins. The six or more surfaces of each individual hepatocyte make contact with adjacent hepatocytes, border the bile canaliculi, or are exposed to the sinusoidal space. Filtered blood travels into the terminal hepatic venules, also called central veins, and then empties into larger hepatic veins and eventually into the inferior vena cava. Functional gradients of hepatocytes based on oxygen saturation have been reported. Hepatocytes closest to the portal triad, which contains the hepatic artery, have greater oxygen saturation than those hepatocytes nearer to the terminal hepatic venule. Blood flows past hepatocytes in zone one, then zone two, and finally zone three before entering the central vein. Hepatocytes in zone one are involved in gluconeogenesis, urea synthesis, and oxidative energy metabolism while those in zone three carry out the functions of glycolysis and lipogenesis.
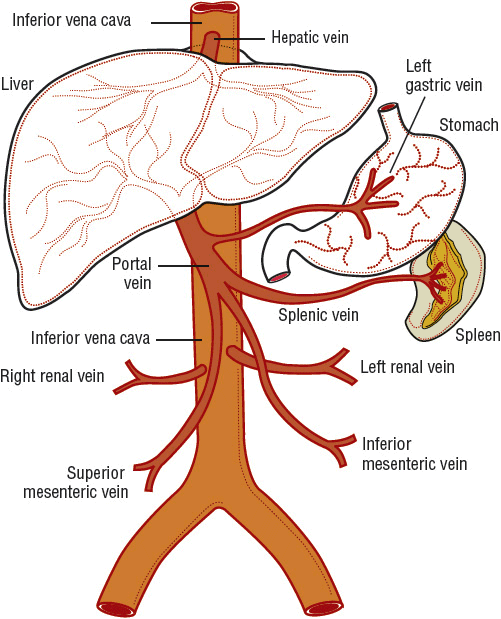
FIGURE 24-1 The portal venous system.
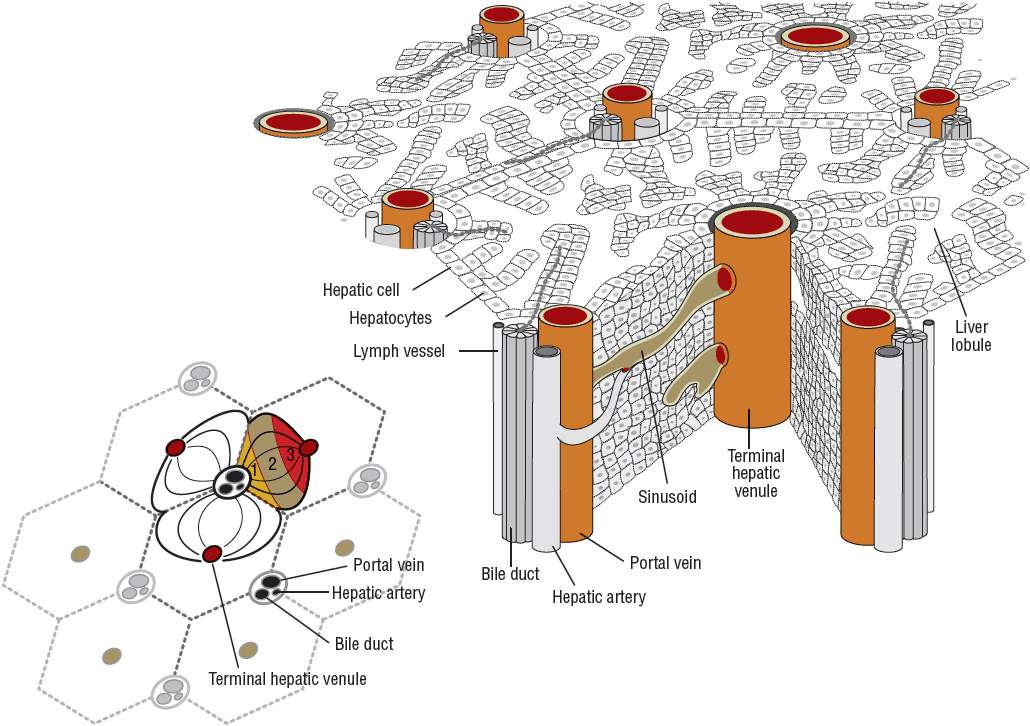
FIGURE 24-2 The hepatic lobule.
Normally, hepatic stellate cells function to store vitamin A and help to maintain the normal matrix in the sinusoidal space.7 During chronic liver disease, however, hepatic stellate cells undergo an “activation” process, which is the central event in the development of hepatic fibrosis. Activation causes stellate cells to lose vitamin A, become highly proliferative, and synthesize fibrotic scar tissue, which accumulates in the sinusoidal space. This leads to loss of hepatocyte microvilli, loss of sinusoidal endothelial fenestrae, deterioration of hepatocyte function, and, if fibrosis progresses, eventual cirrhosis.
Cirrhosis causes changes to the splanchnic vascular bed as well as the systemic circulation.8 Splanchnic vasodilation, decreased responsiveness to vasoconstrictors, and the formation of new blood vessels contribute to an increased splanchnic blood flow, formation of gastroesophageal varices, and variceal bleeding. All of these components are part of the portal hypertensive syndrome. Portal hypertension is characterized by hypervolemia, increased cardiac index, hypotension, and decreased systemic vascular resistance. This is a so-called hyperkinetic syndrome that leads to a marked activation of neurohumoral vasoactive factors, a response that occurs in an effort to maintain the arterial blood pressure within normal limits. Activation of neurohumoral vasoactive factors is a main component in the pathophysiology of the ascites and renal dysfunction that often accompany chronic liver disease. Portal-systemic shunting may also occur and is involved in HE and other complications.
In summary, cirrhosis results from fibrotic changes within the hepatic sinusoids and results in changes in the levels of vasodilatory and vasoconstrictor mediators and an increase in blood flow to the splanchnic vasculature.
ANATOMIC AND PHYSIOLOGIC EFFECTS OF CIRRHOSIS
Cirrhosis and the pathophysiologic abnormalities that cause it result in the commonly encountered problems of ascites, portal hypertension, esophageal varices, HE, and coagulation disorders. Other less commonly seen problems in patients with cirrhosis include hepatorenal syndrome, hepatopulmonary syndrome, and endocrine dysfunction. These are discussed in Management of Portal Hypertension and Variceal Bleeding below.
Ascites
Ascites is the accumulation of an excessive amount of fluid within the peritoneal cavity.9 It is the most commonly occurring major complication of cirrhosis.5 Approximately half of all cirrhotic patients develop ascites within 10 years of diagnosis. Several hypotheses have been offered to explain the mechanism for the development of ascites in decompensated cirrhosis.9 Most acceptable theories state that ascites formation begins as a result of the development of sinusoidal hypertension and portal hypertension. Portal hypertension activates vasodilatory mechanisms that are mediated mostly by nitric oxide overproduction. This leads to splanchnic and peripheral arteriolar vasodilation and, in advanced disease, a drop in arterial pressure. Baroreceptor-mediated activation of the renin–angiotensin–aldosterone system, activation of the sympathetic nervous system, and release of antidiuretic hormone occur in response to the resulting arterial hypotension in an effort to restore normal blood pressure (Fig. 24-3). These changes cause renal sodium and water retention. Additionally, ongoing splanchnic vasodilation increases splanchnic lymph production beyond the capacity of the lymph transportation system. Leakage of lymphatic fluid into the peritoneal cavity occurs. Persistent renal sodium and water retention, increased splanchnic vascular permeability, and lymph leakage into the peritoneal cavity combine to create the sustained ascites formation of end-stage liver disease.
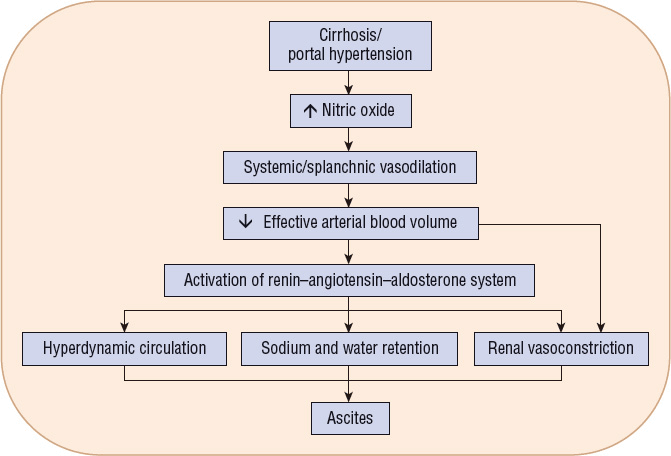
FIGURE 24-3 Pathogenesis of ascites.
Portal Hypertension and Varices
Sinusoidal portal hypertension is most often caused by cirrhosis.10 It is associated with acute variceal bleeding, a medical emergency that carries a mortality rate of 10% to 20% at 6 weeks, and is among the most severe complications of cirrhosis.11 Portal hypertension is defined by the presence of a gradient of greater than 5 mm Hg between the portal and central venous pressures (see Fig. 24-1).10 This gradient is called the hepatic venous pressure gradient (HVPG). Esophageal and gastric varices and variceal bleeding may arise after a HVPG pressure gradient of 10 mm Hg is reached.
Progression to bleeding can be predicted by Child-Pugh score, size of varices, and the presence of red wale markings on the varices. First variceal hemorrhage occurs at an annual rate of about 15% and carries a mortality of 7% to 15%. Rebleeding is common following initial hemorrhage with a median rate of 60% and carries a mortality rate as high as 33%. Prevention of bleeding is a major goal in the therapy of portal hypertension, and strategies include both pharmacologic and surgical approaches.
Hepatic Encephalopathy
HE is a metabolically induced functional disturbance of the brain that is potentially reversible.12 Symptoms of HE are thought to result from an accumulation of gut-derived nitrogenous substances in the systemic circulation as a consequence of decreased hepatic functioning and shunting through portosystemic collaterals bypassing the liver.13 Once these substances enter the CNS, they cause alterations of neurotransmission that affect consciousness and behavior. Ammonia is the most commonly cited culprit in the pathogenesis of HE, but glutamine, benzodiazepine receptor agonists, aromatic amino acids, and manganese are also potential causes.12,13 Arterial ammonia levels are increased commonly in both acute and chronic liver diseases, but an established correlation between blood ammonia levels and mental status does not exist.13 Despite this, interventions to lower blood ammonia levels remain the mainstay of treatment for HE.
HE is now categorized as type A, B, or C based on nomenclature developed by the 11th World Congress of Gastroenterology.12 Type A is HE induced by acute liver failure, type B is due to portal-systemic bypass without associated intrinsic liver disease, and type C is HE that occurs in patients with cirrhosis. Minimal HE refers to cirrhotic patients who do not suffer clinically overt cognitive dysfunction but who are found to have cognitive impairment on psychological studies. The onset of HE in a patient with liver failure may be related to the presence of several known precipitating factors. In cases of HE associated with a precipitant, if that precipitant can be cured or discontinued, it may also be possible to discontinue treatment for HE. In many cases, no precipitant is found and, therefore, long-term treatment of HE may be required.
CLINICAL PRESENTATION Cirrhosis
Coagulation Defects
The liver synthesizes most of the proteins that are responsible for the maintenance of hemostasis (the balance between coagulation and anticoagulation).14 Hepatocellular damage can lead to a disruption in hemostasis because of defects it may cause in the function of coagulation and fibrinolytic factors. These defects include a reduction in the synthesis of clotting factors, excessive fibrinolysis, disseminated intravascular coagulation, thrombocytopenia, and platelet dysfunction. Most coagulation factors are created in the liver, and the levels of these factors can be significantly reduced in chronic liver disease associated with extensive hepatocellular damage. Factor VII is the first factor to decrease as liver function declines due to its short half-life. A reduction in clotting factor VII is common in end-stage liver disease, affecting 60% of patients. Low factor VII activity is prognostic for reduced survival, and the prothrombin time (PT) is a standard component of the Child-Pugh scoring system. Accelerated intravascular coagulation and fibrinolysis can be detected in some patients with cirrhosis. The coexistence of sepsis, shock, surgery, trauma, or ascites may cause a progression from accelerated intravascular coagulation to disseminated intravascular coagulation. In patients with cirrhosis, disseminated intravascular coagulation involves increased release of procoagulants, impaired removal of activated coagulation factors and endotoxins produced by gut bacteria, and reduced synthesis of coagulation inhibitors. Both platelet number and function may also be affected in cirrhosis. Platelet numbers are reduced by multiple mechanisms, including splenomegaly due to portal hypertension and sequestration of platelets in the spleen, reduced hepatic production of thrombopoietin, bone marrow suppression, and increased platelet destruction. Mild to moderate thrombocytopenia occurs in 15% to 70% of patients with cirrhosis. The net effect of the coagulation disorders that occur in cirrhosis is the development of bleeding.
CLINICAL PRESENTATION
Cirrhotic patients may present in a variety of ways, from asymptomatic with abnormal radiographic or laboratory studies to decompensated with ascites, SBP, HE, or variceal bleeding.15
The approach to a patient with suspected liver disease begins with a thorough history and physical exam. Some presenting characteristics of patients with cirrhosis include anorexia, weight loss, weakness, fatigue, jaundice, pruritus, GI bleeding, coagulopathy, increasing abdominal girth with shifting flank dullness, mental status changes, and vascular spiders. Osteoporosis, as a result of vitamin D malabsorption and resultant calcium deficiency, can also occur.
A thorough history including risk factors that predispose patients to cirrhosis should be taken. Quantity and duration of alcohol intake should be determined. Risk factors for hepatitis B and C transmission should be inquired about. These include birthplace in endemic areas, sexual history, intranasal or IV drug use, body piercing or tattooing, and accidental contamination of body tissues or blood. Information concerning any history of transfusions, as well as any personal history of autoimmune or hepatic diseases, should be gathered. A family history should also be taken, looking especially for any family member with a prior history of autoimmune or hepatic diseases.
Laboratory Abnormalities
There are no laboratory or radiographic tests of hepatic function that can accurately diagnose cirrhosis. Despite this, liver function tests, a complete blood count with platelets, and a PT test should be performed if liver disease is suspected. Tests that measure the level of serum liver enzymes are usually referred to as liver function tests.16 However, these tests actually reflect hepatocyte integrity or cholestasis, not liver function.
Routine liver tests include alkaline phosphatase, bilirubin, AST, ALT, and GGT. Additional markers of hepatic synthetic activity include albumin and PT. Liver function tests are often the first step in the evaluation of patients who present with symptoms or signs suggestive of cirrhosis.15 The use of liver function tests in the diagnosis and management of cirrhosis is discussed in the following sections. It may be useful to group the tests into two broad categories: markers of hepatocyte integrity such as the transaminases and markers of liver function mass such as PT and albumin.16
Aminotransferases
The aminotransferases, AST and ALT, are enzymes that are highly concentrated in the liver. Liver injury, whether acute or chronic, results, at some point in the course of the disease, in increases in the serum concentrations of the aminotransferase enzymes. The degree of elevation, rate of rise, and nature of the course of alteration in aminotransferase serum levels are helpful in suggesting possible etiologies. Liver function tests will typically be elevated to the highest levels in acute viral, ischemic, or toxic liver injury. Chronic hepatitis and cirrhosis patients may present with elevated aminotransferase levels, but they may also present with aminotransferase levels within the normal reference range. The degree of aminotransferase level elevation is dependent on the course of the hepatic injury being experienced by the patients and also depends on when the enzyme levels are tested. In a landmark study by Cohen and Kaplan, alcoholic liver disease resulted in AST elevations of only six to seven times the upper limit of normal in 98% of patients.17 The ratio of AST to ALT also provides information in patients with suspected alcoholic liver disease. Seventy percent of patients with alcoholic liver disease in the study by Cohen and Kaplan had ratios greater than 2, whereas 92% of patients had ratios greater than 1.
Alkaline Phosphatase and γ-Glutamyl Transpeptidase
Elevated serum levels of alkaline phosphatase and GGT occur in cases of liver injury with a cholestatic pattern and therefore accompany conditions such as primary biliary cirrhosis, primary sclerosing cholangitis, drug-induced cholestasis, bile duct obstruction, autoimmune cholestatic liver disease, and metastatic cancer of the liver.16 Neither alkaline phosphatase nor GGT is found solely in the liver, and elevations in either of these biomarkers can occur in a variety of disease states affecting other bodily tissues. However, the combination of an elevation in alkaline phosphatase level with a concomitant elevation in GGT level increases clinical suspicion of hepatic etiology.
Child-Pugh Classification and Model for End-Stage Liver Disease Score
The Child-Pugh classification system has gained widespread acceptance as a means of quantifying the myriad effects of the cirrhotic process on the laboratory and clinical manifestations of this disease.18 Recommended drug dosing adjustments for patients in liver failure, when available, are normally based on the Child-Pugh score. The newer Model for End-Stage Liver Disease (MELD) scoring system is now the accepted classification scheme used by the United Network for Organ Sharing in the allocation livers for transplantation.19 The Child-Pugh classification system employs a combination of physical and laboratory findings (Table 24-2), whereas the MELD score calculation takes into account a patient’s serum creatinine, bilirubin, international normalized ratio (INR), and etiology of liver disease, omitting the more subjective reports of ascites and encephalopathy used in the Child-Pugh system. The MELD scoring calculation* is as follows20:
TABLE 24-2 Criteria and Scoring for the Child-Pugh Grading of Chronic Liver Disease
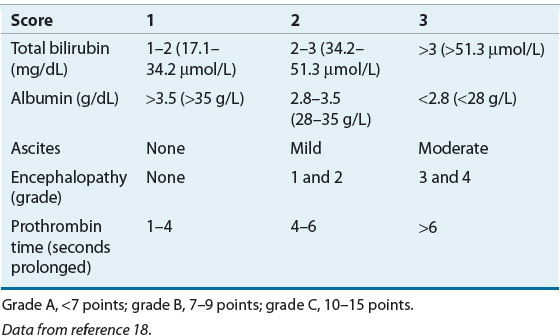
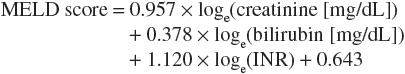
or using SI units*:

These classification systems are important because they are used to assess and define the severity of the cirrhosis, and as a predictor for patient survival, surgical outcome, and risk of variceal bleeding.
Bilirubin
Bilirubin is the product of the breakdown of hemoglobin molecules in the reticuloendothelial system.16 Elevations in serum conjugated bilirubin indicate that the liver has lost at least half of its excretory capacity and are usually a sign of liver disease. When found in conjunction with markedly elevated AST and ALT, conjugated hyperbilirubinemia indicates the possible presence of acute viral hepatitis, autoimmune hepatitis, toxic liver injury, or ischemic liver injury. Elevated conjugated bilirubin levels with concomitant increases in alkaline phosphatase and normal aminotransferase levels are a sign of cholestatic disease and possible cholestatic drug reactions. Causes of elevations in unconjugated bilirubin include hemolysis, Gilbert’s syndrome, hematoma reabsorption, and ineffective erythropoiesis. Causes of conjugated hyperbilirubinemia include bile duct obstruction, hepatitis, cirrhosis, primary sclerosing cholangitis, primary biliary cirrhosis, total parenteral nutrition, drug toxins, and vanishing bile duct syndrome. When cirrhosis has been established, the degree of bilirubin elevation has prognostic significance and is used as a component of the Child-Pugh and MELD scoring systems for quantifying the degree of cirrhosis.18,20
Figure 24-4 describes a general algorithm for the interpretation of liver function tests. The algorithm first separates the tests into two categories based on the underlying pathology (pattern of elevations): obstructive (alkaline phosphatase, GGT, and bilirubin) versus hepatocellular (AST and ALT). If a hepatocellular pattern predominates, the magnitude of elevation provides diagnostic assistance. If the degree of elevation is greater than 10 times normal, the etiology is likely a result of drugs or other toxins, ischemia, or acute viral hepatitis.16 Elevations less than 10 times normal have a broad differential. Unfortunately, most liver enzyme abnormalities will fall into a mixed pattern providing limited diagnostic assistance.
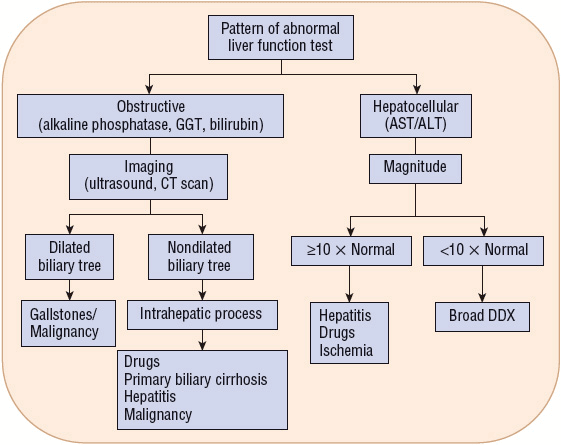
FIGURE 24-4 Interpretation of liver function tests. (DDX, differential diagnosis.)
Albumin and Coagulation Factors
Albumin and coagulation proteins are markers of hepatic synthetic activity and are therefore used to estimate the level of hepatic functioning in cirrhosis. Albumin and PT are used in the Child-Pugh system for quantifying liver disease, and the INR is used in the MELD scoring system as a marker of coagulation.18,20 Albumin levels can be affected by a number of factors, including malnutrition, malabsorption, and protein losses from renal and intestinal sources.16
Coagulation factors I, II, V, VII, VIII, IX, X, XI, XII, and XIII are synthesized in the liver.14 Significantly reduced levels of coagulation factors II, V, VII, and XIII have been observed in patients with chronic liver disease resulting in PT prolongation and systemic bleeding unrelated to portal hypertension.
Thrombocytopenia
Thrombocytopenia (generally defined as a platelet count less than 150,000/mm3 [150 × 109/L]) is a common feature of chronic liver disease found in 15% to 70% of cirrhotic patients depending on the stage of liver disease and definition of thrombocytopenia. The etiology of thrombocytopenia in liver disease is multifactorial, involving primarily splenomegaly due to portal hypertension with pooling of platelets in the spleen. A decrease in thrombopoietin due to decreased hepatic synthesis occurs as well as an immune-mediated destruction of platelets. Additionally, bone marrow suppression related to the hepatitis C virus or interferon antiviral treatment may exist and lead to thrombocytopenia associated with the cirrhotic process.
Endoscopic and Radiographic Abnormalities
While no radiographic test is considered a diagnostic standard for cirrhosis, radiographic studies may be used to detect ascites, hepatosplenomegaly, hepatic or portal vein thromboses, and hepatocellular carcinoma.15 Ultrasonography, because it does not require radiation exposure or IV contrast and is relatively low cost, should be the first radiographic study in the evaluation of a patient with suspected cirrhosis. Hepatic nodularity, irregularity, increased echogenicity, and atrophy are all ultrasonographic findings indicative of cirrhosis. Ascites may also be detected on ultrasound. Computed tomography and magnetic resonance imaging can demonstrate liver nodularity as well as atrophic and hypertrophic changes. Ascites and varices may also be detected on computed tomography or magnetic resonance imaging scans. Portal vein patency can be assessed by computer tomography imaging.
Liver Biopsy
Liver biopsy should be considered after a thorough noninvasive workup has failed to confirm a diagnosis in suspected cirrhosis. Liver biopsy has a sensitivity and specificity of 80% to 100% for an accurate diagnosis of cirrhosis and its etiology. The success of biopsy as a diagnostic tool is dependent on the number of histologic samples retrieved as well as the sampling method used.
TREATMENT
General Approaches to Treatment
General approaches to therapy in cirrhosis should include the following:
1. Identify and eliminate, where possible, the causes of cirrhosis (e.g., alcohol abuse).
2. Assess the risk for variceal bleeding and begin pharmacologic prophylaxis when indicated. Prophylactic endoscopic therapy can be used for patients with high-risk medium and large varices as well as in patients with contraindications or intolerance to nonselective β-adrenergic blockers. Endoscopic therapy is also appropriate for patients suffering acute bleeding episodes. Variceal obliteration with endoscopic techniques in conjunction with pharmacologic intervention is the recommended treatment of choice in patients with acute bleeding.
3. Evaluate the patient for clinical signs of ascites and manage with pharmacologic therapy (e.g., diuretics) and paracentesis. Careful monitoring for SBP should be used in patients with ascites who undergo acute deterioration.
4. HE is a common complication of cirrhosis and requires clinical vigilance and treatment with dietary restriction, elimination of CNS depressants, and therapy to lower ammonia levels.
5. Frequent monitoring for signs of hepatorenal syndrome, pulmonary insufficiency, and endocrine dysfunction is necessary.
Desired Outcomes
The desired therapeutic outcomes can be viewed in two categories: resolution of acute complications such as tamponade of bleeding and resolution of hemodynamic instability for an episode of acute variceal hemorrhage and prevention of complications through lowering of portal pressure with medical therapy using β-adrenergic blocker therapy or supporting abstinence from alcohol. Treatment end points and desired therapeutic outcomes are presented for each of the recommended therapies discussed.
Management of Portal Hypertension and Variceal Bleeding
The management of varices involves three strategies: (a) primary prophylaxis (prevention of the first bleeding episode); (b) treatment of acute variceal hemorrhage; and (c) secondary prophylaxis (prevention of rebleeding in patients who have previously bled).11
Primary Prophylaxis
β-Adrenergic Blockade The mainstay of primary prophylaxis is the use of nonselective β-adrenergic blocking agents such as propranolol or nadolol.10,11,21 These agents reduce portal pressure by reducing portal venous inflow via two mechanisms: a decrease in cardiac output through β1-adrenergic blockade and a decrease in splanchnic blood flow through β2-adrenergic blockade.10
Endoscopic Variceal Ligation (EVL) EVL is an endoscopic therapy that consists of placing rubber bands around varices until the varices are obliterated.21
Treatment Recommendations: Variceal Bleeding—Primary Prophylaxis
![]() All patients with cirrhosis should be screened for varices on diagnosis.10,11,21 β-Adrenergic blocker therapy is not indicated in patients without varices to prevent the formation of varices. Patients with small varices plus risk factors for variceal hemorrhage including red wale marks or Child-Pugh class C should receive prophylaxis therapy with a nonselective β-adrenergic blocker. β-Adrenergic blocker therapy is recommended preferentially to EVL in this situation due to the technical difficulty of EVL in the treatment of small varices. β-Adrenergic blocker therapy is not recommended for patients with small varices in the absence of risk factors as there is insufficient evidence to support this therapy to slow the growth of varices in this scenario. All patients found to have medium to large varices that have not bled should receive primary prophylaxis therapy with a nonselective β-adrenergic blocker or EVL. The choice of treatment should be based on a consideration of resources and expertise as well as patient preferences and characteristics with a particular emphasis on side effects and contraindications.11 If β-adrenergic blocker therapy is chosen, initiate therapy with oral propranolol 20 mg twice daily or nadolol 20 to 40 mg once daily and titrate every 2 to 3 days to maximal tolerated dose to heart rates of 55 to 60 beats/min.10,21 Once a patient is started on nonselective β-adrenergic blocker therapy, it should be continued indefinitely. Following initiation and appropriate titration of the β-adrenergic blocker, further endoscopic surveillance is not needed. If EVL is chosen, it will be performed every 1 to 2 weeks until the obliteration of varices.21 Followup surveillance will occur at 1 to 3 months and again every 6 to 12 months thereafter.
All patients with cirrhosis should be screened for varices on diagnosis.10,11,21 β-Adrenergic blocker therapy is not indicated in patients without varices to prevent the formation of varices. Patients with small varices plus risk factors for variceal hemorrhage including red wale marks or Child-Pugh class C should receive prophylaxis therapy with a nonselective β-adrenergic blocker. β-Adrenergic blocker therapy is recommended preferentially to EVL in this situation due to the technical difficulty of EVL in the treatment of small varices. β-Adrenergic blocker therapy is not recommended for patients with small varices in the absence of risk factors as there is insufficient evidence to support this therapy to slow the growth of varices in this scenario. All patients found to have medium to large varices that have not bled should receive primary prophylaxis therapy with a nonselective β-adrenergic blocker or EVL. The choice of treatment should be based on a consideration of resources and expertise as well as patient preferences and characteristics with a particular emphasis on side effects and contraindications.11 If β-adrenergic blocker therapy is chosen, initiate therapy with oral propranolol 20 mg twice daily or nadolol 20 to 40 mg once daily and titrate every 2 to 3 days to maximal tolerated dose to heart rates of 55 to 60 beats/min.10,21 Once a patient is started on nonselective β-adrenergic blocker therapy, it should be continued indefinitely. Following initiation and appropriate titration of the β-adrenergic blocker, further endoscopic surveillance is not needed. If EVL is chosen, it will be performed every 1 to 2 weeks until the obliteration of varices.21 Followup surveillance will occur at 1 to 3 months and again every 6 to 12 months thereafter.
Patients with contraindications to therapy with nonselective β-adrenergic blockers (i.e., those with asthma, insulin-dependent diabetes with episodes of hypoglycemia, and peripheral vascular disease) or intolerance to β-adrenergic blockers should be considered for alternative prophylactic therapy with EVL.22 Also, EVL may be considered as a possible first option for primary prophylaxis in patients with high-risk medium to large varices. Nitrates are no longer recommended as alternative therapy for primary prophylaxis against variceal bleeding in patients with intolerance to nonselective β-adrenergic blocker due to a potential for higher mortality with this therapy.21 At this time, there is also insufficient evidence to support the use of other therapies and procedures (such as combination nonselective β-adrenergic blocker therapy with isosorbide mononitrate, combination nonselective β-adrenergic blocker therapy with spironolactone, combination nonselective β-adrenergic blocker therapy with EVL, shunt surgery, and endoscopic sclerotherapy) for primary prevention of variceal hemorrhage.
Acute Variceal Hemorrhage
Variceal hemorrhage is a medical emergency that carries a mortality rate of 15% to 20%, requires admission to an intensive care unit, and is one of the most feared complications of cirrhosis.10,21 Treatment of acute variceal bleeding includes general stabilizing and assessment measures as well as specific measures to control the acute hemorrhage and prevent complications.
Initial treatment goals include (a) adequate blood volume resuscitation, (b) protection of airway from aspiration of blood, (c) correction of significant coagulopathy and/or thrombocytopenia with fresh-frozen plasma and platelets, (d) prophylaxis against SBP and other infections, (e) control of bleeding, (f) prevention of rebleeding, and (g) preservation of liver function.22 Prompt stabilization of blood volume with a goal of maintaining hemodynamic stability and a hemoglobin of 8 g/dL (80 g/L; 4.97 mmol/L) should be undertaken. Volume should be expanded to maintain a systolic blood pressure of 90 to 100 mm Hg and a heart rate of less than 100 beats/min, but vigorous resuscitation with saline solution should generally be avoided because this may lead to recurrent variceal hemorrhage or accumulation of ascites and/or fluid at other anatomic sites.21,22 Use of recombinant factor VIIa therapy is not recommended in cirrhotic patients with GI hemorrhage at this time. Airway management is critical in patients with variceal hemorrhage, especially those with concomitant HE or severe bleeding.22 Elective or more emergent intubation may be required prior to diagnostic endoscopy. Combination pharmacologic therapy plus endoscopic therapy with preferably EVL, or sclerotherapy if EVL is not technically feasible, is considered the most rational approach to the treatment of acute variceal bleeding.10,21
Vasoactive drug therapy (usually octreotide) is routinely used early to stop or slow bleeding for patient management as soon as a diagnosis of variceal bleeding is suspected, and potentially even before endoscopy. Antibiotic therapy to prevent SBP and other infections, as well as to prevent rebleeding and decrease mortality, should be implemented. Figure 24-5 presents an algorithm for the management of variceal hemorrhage.
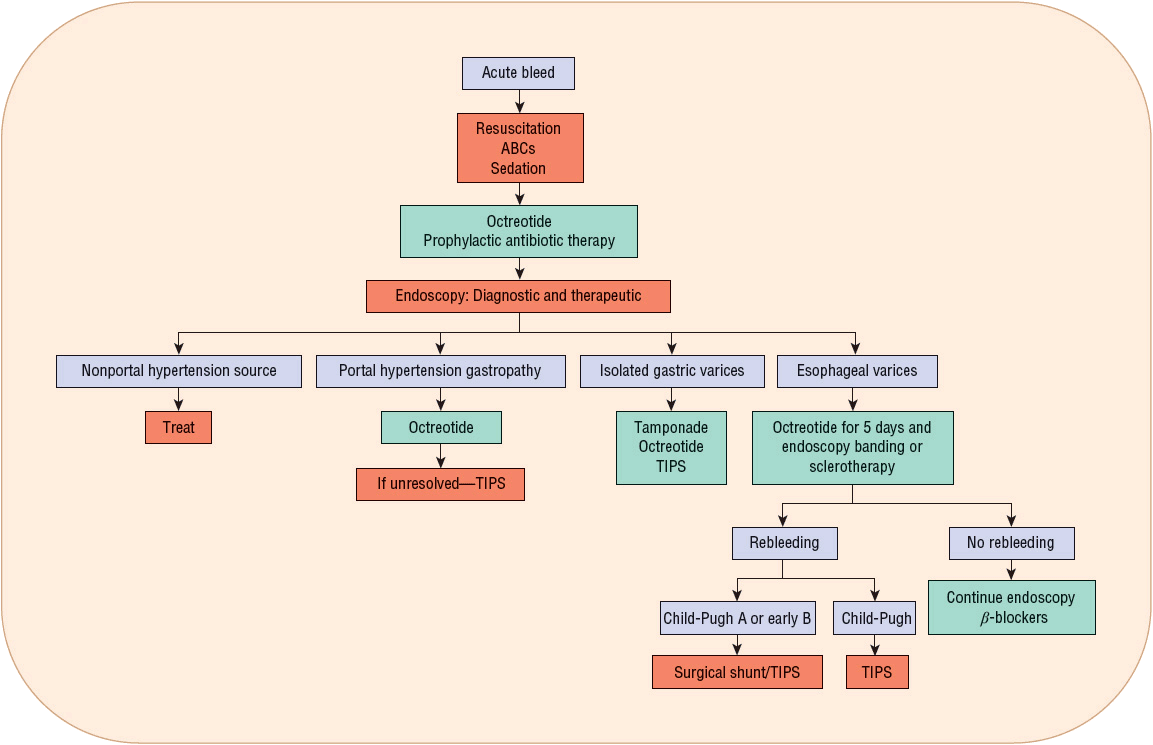
FIGURE 24-5 Management of acute variceal hemorrhage.



