Poorly Differentiated Carcinoma
Yuri E. Nikiforov
DEFINITION
Poorly differentiated thyroid carcinoma is an aggressive malignant tumor of follicular cell origin that is characterized by a partial loss of thyroid differentiation and occupies morphologically and behaviorally an intermediate position between well-differentiated papillary and follicular carcinomas and fully dedifferentiated anaplastic carcinoma. Another term frequently used to designate this tumor is insular carcinoma, although its usage should be discouraged as it places an emphasis on the growth pattern, which is not specific for this tumor type, rather than on the cell morphology.
HISTORICAL COMMENTS AND EVOLUTION OF DIAGNOSTIC CRITERIA
The tumor currently known as poorly differentiated carcinoma was first described by Sakamoto and colleagues in 19831 and by Carcangiu and colleagues in 1984.2 In the first report, Sakamoto et al. analyzed a group of 258 thyroid tumors from Japan and found that those with solid, trabecular, or scirrhous (sclerotic) growth pattern demonstrated biologic behavior intermediate between the rest of the papillary and follicular carcinomas on one side and anaplastic thyroid carcinoma on another. They called tumors with this architecture poorly differentiated carcinoma, irrespective of other histologic characteristics of tumor cells. In the article by Carcangiu et al., the authors reported a series of 25 cases of unusual thyroid carcinoma from the University of Florence Medical School in Italy. These tumors formed characteristic solid nests or “insulae” and were characterized by small size and uniformity of cells, significant mitotic activity, capsular and vascular invasion, and tumor necrosis. The authors postulated that morphologically and behaviorally, this tumor occupied an intermediate position between well-differentiated papillary and follicular carcinoma and anaplastic carcinoma and named it poorly differentiated or insular carcinoma. They noted that this tumor resembled one that was described by Langhans in 1907 as “wuchernde Struma” (“proliferating struma”). These two observations established a concept of thyroid tumor of intermediate differentiation and behavior, but offered significantly different histologic criteria for its diagnosis. Sakamoto and colleagues based the diagnosis exclusively on tumor growth pattern, whereas Carcangiu and colleagues described a distinctive growth pattern as well as other morphologic characteristics of tumor cells, albeit with no clear indication of what features are sufficient to establish the diagnosis.
Subsequent reports have used either the more broad Sakamoto criteria, adding other unusual variants of papillary carcinoma, such as columnar cell variant, diffuse sclerosing variant, and even tall cell variant to the category of poorly differentiated carcinoma,3,4,5 or the more restrictive Carcangiu criteria, but still emphasizing an insular growth pattern as sufficient criterion alone for inclusion into the category of poorly differentiated carcinoma.6,7,8 As a result, various groups reported series of tumors carrying the same name but, in reality, having quite different morphologic characteristics. For the most part, the criteria used were broad, which resulted in frequent inclusion in this category some well-differentiated carcinomas, such as solid variant of papillary carcinoma. The 2004 World Health Organization classification of thyroid tumors recognized poorly differentiated carcinoma as a specific entity, stating that it is characterized by solid, trabecular, or insular architecture; infiltrative growth; necrosis; and vascular invasion.9 Although this description included features beyond the growth pattern, it did not specify how many of those were sufficient to establish this diagnosis. Still some authors suggested that poorly differentiated carcinoma has to be defined exclusively based on the presence of necrosis and high mitotic activity, as these features better stratified patients into prognostic categories than a growth pattern.10
In 2006, an international working group of thyroid pathologists from Europe, Japan, and the United States, which included the principal authors of the two original publications, offered a set of consensus diagnostic criteria for this tumor, which was developed at a meeting hosted by Dr. Gianni Bussolati in Turin, Italy.11 The unifying diagnostic criteria included both the architectural pattern and the cytologic features of tumor cells and were developed based on a series of tumors submitted from different countries and reviewed by a panel of 12 thyroid pathologists. These criteria showed good correlation with tumor behavior and are relatively simple and expected to be reproducible between observers. The Turin consensus criteria provide the basis for discussion in this chapter. As these criteria are relatively recent and many observations available in the literature used various selection criteria, this chapter primarily incorporates the results of studies that used either Turin criteria or reasonably restrictive criteria to define poorly differentiated thyroid carcinoma.
INCIDENCE AND EPIDEMIOLOGY
Poorly differentiated thyroid carcinoma is a rare tumor. When defined by the Turin diagnostic criteria, its incidence varies from <1% in Japan12 to 1.8% in the United States.13 Some geographic areas, however, may have a higher incidence of this tumor. Specifically, in the Piemonte region of northern Italy, poorly differentiated carcinoma historically accounted for 4% to 7% of all malignant thyroid tumors,6,14 and the incidence remained at 6.7% when the Turin diagnostic criteria were applied.13
Poorly differentiated carcinoma typically presents on average one decade later than well-differentiated carcinomas. In most series, the mean age of patient was between 55 and 63 years.1,2,6,11,13 Poorly differentiated carcinoma is exceedingly rare in children and young adults, and this diagnosis should be established with great caution in individuals younger than 30 years. The youngest age of patients in most reported series was 33 to 34 years,2,11 although it was 14.1 years in one recent study.13 The tumor is more common in females, with a female predilection ranging from 1.6 to 2:1 in large series of patients.1,2,11,13
ETIOLOGIC FACTORS
The etiology remains unknown. Some tumors develop from preexisting well-differentiated papillary or follicular carcinoma, whereas others are likely to develop de novo. The association with benign thyroid disease is suspected based on presumably higher prevalence of this tumor in the Piemonte region of Italy located in the Alps, which used to be an area of endemic goiter.6,14 In this region, most patients with poorly differentiated carcinoma had a history of goiter, which was confirmed in almost all thyroids at surgery. A history of exposure to therapeutic radiation to the neck region was found in 12% of patients in one series,2 although this association remains unconfirmed.
PATHOGENESIS AND MOLECULAR GENETICS
It is generally accepted that poorly differentiated carcinoma may develop through three pathogenetic pathways: (1) by partial dedifferentiation of papillary carcinoma, (2) by partial dedifferentiation of follicular (including oncocytic type) carcinoma, and (3) de novo, without a preexisting well-differentiated carcinoma precursor (Fig. 12.1). This assumption is supported by frequent finding of a well-differentiated papillary or follicular carcinoma intermixed with poorly differentiated carcinoma areas (Fig. 12.2) and by observations of temporal progression from classic papillary carcinoma to poorly differentiated carcinoma in subsequent tumor recurrences.15 In a recent series of 152 tumors, 34% of them did not have a well-differentiated component, whereas the rest showed a residual well-differentiated papillary or follicular carcinoma.13
Somatic Mutations
Somatic mutations that occur in poorly differentiated carcinoma are summarized in Table 12.1. Generally, they can be divided into two groups: (1) mutations that also occur in the well-differentiated tumor component and therefore represent an early event in tumorigenesis, initiating the development of well-differentiated cancer and predisposing to subsequent additional molecular events that govern tumor dedifferentiation and (2) mutations that are present only in the poorly differentiated tumor component and therefore are late events, directly driving the process of dedifferentiation (Fig. 12.1). The first group includes BRAF and RAS mutations. The second group includes TP53 and β-catenin (CTNNB1) mutations, which occur in poorly differentiated and also anaplastic carcinomas but not in well-differentiated cancers. Mutations in the effectors of the PI3K/PTEN/AKT signaling pathway are more difficult to place in one of these groups, although they are more likely to represent late events. Of note, clonal RET/PTC and PAX8/PPARγ rearrangements are less commonly found in poorly differentiated (and anaplastic) carcinomas,16 suggesting that these oncogenes are less likely to create a “molecular environment” that promotes tumor dedifferentiation. Some studies, however, have reported a more common (18%) occurrence of RET/PTC rearrangement in poorly differentiated carcinomas defined on the basis of tumor necrosis and high mitotic activity.17
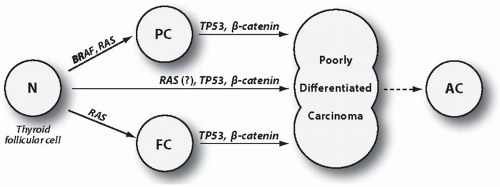 FIGURE 12.1. Poorly differentiated carcinoma may develop by partial dedifferentiation of well-differentiated papillary carcinoma (PC) or follicular carcinoma (FC), or directly, de novo, without a well-differentiated carcinoma stage. The process of dedifferentiation can proceed further and result in transformation to anaplastic carcinoma (AC). BRAF and RAS mutations are found in both well-differentiated and poorly differentiated carcinomas, indicating that they occur early and confer well-differentiated carcinoma cells with propensity to undergo dedifferentiation, most likely by acquiring additional mutations, such as those of TP53 and β-catenin (CTNNB1) genes. PC, papillary carcinoma; FC, follicular carcinoma; AC, anaplastic carcinoma. |
RAS Mutations
Activating point mutations of the RAS genes are found in 20% to 40% of poorly differentiated carcinomas.13,17,18,19 The most commonly affected hot spot is NRAS codon 61, followed by HRAS codon 61. RAS mutations are not restricted to this tumor type because they can also occur in follicular adenomas, follicular carcinomas, and follicular variant of papillary carcinomas. Many poorly differentiated carcinomas with a RAS mutation contain an adjacent component of well-differentiated follicular carcinoma or follicular variant of papillary carcinoma. When areas of poorly differentiated carcinoma and well-differentiated carcinoma are microdissected separately for DNA extraction, an identical RAS mutation is found in both components, confirming that it is an early event (Fig. 12.3).
Mutant RAS activates various downstream signaling pathways that stimulate cell proliferation and survival, most important of which in thyroid cells are mitogen-activated protein kinase (MAPK) and PI3K/AKT pathways. Some experimental data suggest that mutant RAS may interfere with DNA damage response and promote chromosome instability.20,21 The increasing instability
would stimulate the acquisition of additional mutations, which could in turn initiate the process of tumor dedifferentiation. However, RAS mutation on its own is unlikely to be sufficient to drive tumor dedifferentiation, as it is common in well-differentiated carcinomas and even in benign thyroid adenomas.
would stimulate the acquisition of additional mutations, which could in turn initiate the process of tumor dedifferentiation. However, RAS mutation on its own is unlikely to be sufficient to drive tumor dedifferentiation, as it is common in well-differentiated carcinomas and even in benign thyroid adenomas.
BRAF Mutations
BRAF mutations, which are a characteristic feature of papillary carcinomas, also occur in poorly differentiated carcinomas. The reported incidence of this mutation varies substantially and on average is approximately 15%.17,19,22,23 Most BRAF-positive poorly differentiated carcinomas contain a well-differentiated papillary carcinoma component, typically a tall cell variant (Fig. 12.2A). Both tumor components reveal a V600E BRAF mutation when studied separately, indicating that it occurs early in carcinogenesis.22 In one study, BRAF mutations were found in 12% of the entire series of poorly differentiated carcinoma but in 47% of more aggressive, radioactive iodine refractory and [18F]fluorodeoxyglucose positron emission tomography (FDG-PET) positive poorly differentiated carcinomas.17
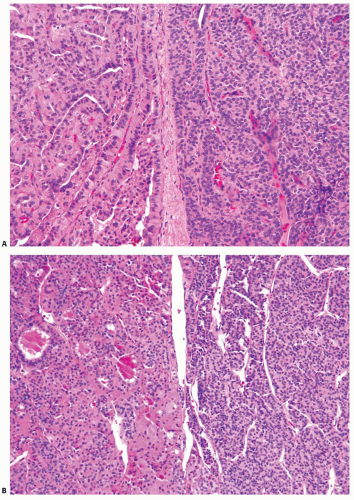 FIGURE 12.2. Poorly differentiated carcinoma (right side of both panels) coexisting with papillary carcinoma, tall cell variant (A), and with oncocytic follicular carcinoma (B). |
The oncogenic qualities of BRAF V600E mutation are associated with activation of the MAPK signaling pathway. Chronic overstimulation of this pathway by mutant BRAF in thyroid cells of transgenic mice results in the formation of papillary carcinomas that in time undergo progression to poorly differentiated carcinoma.24 Similar to human poorly differentiated carcinomas, the poorly differentiated foci in these animals showed solid sheets
of cells with more uniform nuclei, scant cytoplasm, and loss of nuclear features of papillary carcinoma (Fig. 12.4). The process of dedifferentiation in the transgenic animals was associated with profound dysregulation of expression of genes involved in cell adhesion and intracellular junction, providing evidence for epithelial-mesenchymal transition in these tumors.25
of cells with more uniform nuclei, scant cytoplasm, and loss of nuclear features of papillary carcinoma (Fig. 12.4). The process of dedifferentiation in the transgenic animals was associated with profound dysregulation of expression of genes involved in cell adhesion and intracellular junction, providing evidence for epithelial-mesenchymal transition in these tumors.25
Table 12.1 Average Prevalence of Mutations in Poorly Differentiated Thyroid Carcinoma and Other Thyroid Carcinomas | ||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||
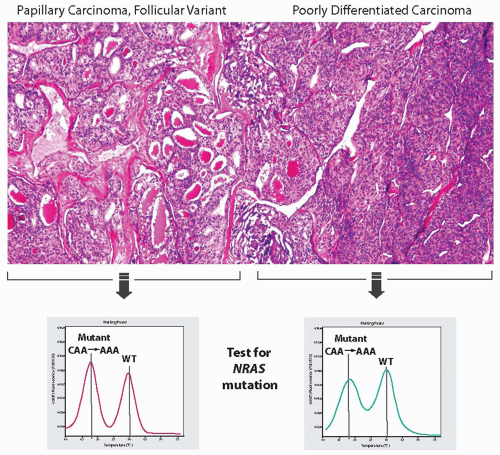 FIGURE 12.3. Poorly differentiated carcinoma coexisting with the follicular variant of papillary carcinoma. DNA was isolated separately from the two tumor components and subjected to testing for RAS mutations by real-time PCR and post-PCR melting curve analysis on LightCycler, which revealed an identical CAA → AAA NRAS mutation at codon 61 in both tumor components. |
TP53 Mutations
Mutations of the TP53 tumor suppressor gene occur in about 30% of poorly differentiated carcinomas.26,27,28 In contrast to RAS and BRAF, these mutations are very rare in well-differentiated thyroid carcinomas and represent a late event in thyroid carcinogenesis, being most prevalent in anaplastic thyroid carcinomas.
The TP53 gene encodes a nuclear transcription factor that plays a central role in the regulation of the cell cycle, DNA repair, and apoptosis. It exerts these functions by its ability to transactivate the expression of genes coding for cell cycle proteins such as p21/WAF1. Mutations typically affect exons 5 to 8 of TP53 and include inactivating point mutations, small deletions, or insertions. Inactivation of TP53 function results in progressive genome destabilization, accumulation of additional mutations, and emergence of more malignant and less differentiated tumor clones.29,30 A sharply increased incidence of TP53 mutations from well-differentiated to poorly differentiated and then to anaplastic carcinomas suggests that TP53 inactivation is crucial for stepwise thyroid cancer progression and plays a direct role in triggering tumor dedifferentiation.
β-Catenin (CTNNB1) Mutations
β-Catenin mutation is another late event potentially involved in tumor dedifferentiation. This cytoplasmic protein, encoded by the CTNNB1 gene, plays an important role in cell adhesion and signaling along the wingless (Wnt) pathway. Normally, in the absence of Wnt signaling, the protein is located at the inner surface of the cell membrane and at a low level in the cytoplasm, where it is rapidly degraded by the adenomatous polyposis coli (APC) multiprotein complex. Wnt binding stabilizes the protein that accumulates in the cytoplasm and translocates to the nucleus, where it upregulates the transcriptional activity of different genes. Point mutations in exon 3 of CTNNB1 stabilize the protein by making it insensitive for APC-induced degradation. This results in the accumulation of β-catenin in the nucleus and constitutive activation of the target gene expression.
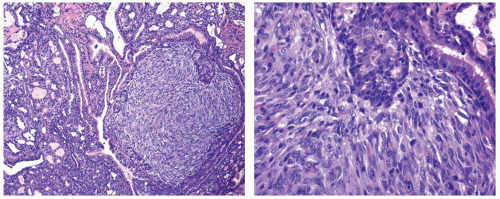 FIGURE 12.4. Microscopic appearance of thyroid tumors developed in transgenic mice with thyroid-specific expression of mutant BRAF V600E. This papillary thyroid carcinoma has tall cell features and focal areas of poorly differentiated carcinoma that are visible as a sharply demarcated solid area showing a uniform population of cells with elongated nuclei and reduced amount of cytoplasm. Based on the data reported by Knauf et al.24
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|