ETIOLOGY
Nearly all cases of thyroid gland lymphoma arise in the setting of chronic lymphocytic (Hashimoto) thyroiditis.
3,
4,
7,
10,
29,
30,
31,
32,
33,
34,
35,
36,
37,
38 In fact, the estimated relative risk of developing a lymphoma is 67 to 80 in patients with chronic lymphocytic thyroiditis when compared with age- and sex-matched controls.
39,
40 Chronic lymphocytic thyroiditis shows an infiltrate of lymphoid cells that can be nodular or diffuse, frequently associated with lymphoid follicle formation including germinal centers (GCs), fibrosis, and oncocytic metaplasia of the thyroid follicular epithelial cells (see
Chapter 4). Fibrosis and squamous metaplasia frequently accompany the lymphoid infiltrate. The presence of serologic antithyroid autoantibodies is requisite for the diagnosis of Hashimoto thyroiditis. Three possible processes are postulated for the development of acquired MALT: an autoimmune process, an immune deficiency, or an inflammatory process. Although
Chlamydia psittaci DNA has been identified in a subset of thyroid MALT lymphoma,
41 the etiologic agent responsible for chronic antigenic stimulation may not be known in all cases. Similar to their gastrointestinal, salivary gland, and lacrimal counterparts, there may be progression from a polyclonal, antigen-driven response to a monoclonal proliferation and subsequent development into an overt lymphoma. Interestingly, thyroid lymphomas may also show an increased ratio of CD8+ cells (suppressor/cytotoxic cell) to CD4+ cells (helper/inducer cell) as compared with lymphocytic thyroiditis alone, providing support for a difference in local immunologic conditions.
29,
42
PATHOGENESIS AND MOLECULAR GENETICS
Chronic lymphocytic thyroiditis is almost certainly a requisite for the development of lymphoma in the thyroid gland. Atrophy of the residual thyroid parenchyma and fibrosis supports the chronicity of the underlying process (acquired MALT) that is associated with the subsequent development of lymphoma.
The cytogenetic and molecular genetic features of MALT lymphoma of the thyroid gland have not been as extensively studied as in other sites. There seem to be anatomic site specific chromosomal frequencies, but three MALT lymphoma-associated translocations [t(11;18)(q21;q21), t(1;14)(p22;q32), and t(14;18) (q32;q21)] result in the constitutive activation of the nuclear factor-
κβ oncogenic pathway.
43,
44 The t(11;18)(q21;q21) results in a chimeric fusion of the
API2 region on chromosome 11q21 with the
MALT1 gene on chromosome 18q21, yielding a product that may concomitantly work as a tumor suppressor gene and as an oncogene.
45 Although present in a large proportion of gastrointestinal and lung MALT lymphomas (24% to 53%), the t(11;18)(q21;q21) translocation is only rarely reported in thyroid lymphomas.
46,
47,
48,
49,
50 The t(3;14)(p14.1;q32) involving
IGH and the forkhead box protein P1 (
FOXP1) has been reported in up to 50% of thyroid MALT lymphoma and seems to be mutually exclusive of t(1;14), t(14;18), and t(11;18) rearrangements but may be accompanied by trisomy 3 or other aneuploidies.
50,
51 The molecular mechanisms of this translocation and significance of deregulation of
FOXP1 expression have not been fully elucidated.
Microsatellite instability and loss of heterozygosity are not identified in thyroid lymphoma. Aberrant
p15,
p16, and
p73 promoter methylation is quite common.
36 TP53 mutation followed by complete inactivation by the loss of the second allele may be associated with high-grade transformation. It is suggested that CD40 signaling in combination with Th2 cytokines is necessary for the development and progression of low-grade MALT lymphoma. T cells, which activate B cells in a CD40-dependent fashion, may contribute to lymphoma pathogenesis and may be identified in lymphocytic thyroiditis.
52 Epstein-Barr virus has been detected in thyroid lymphoma, but in a very limited number of cases, suggesting it is not a major etiologic agent.
53Several theories have been proposed about the putative cell of origin for MALT lymphoma. Carcinogenesis is a multistep, multifactorial process involving the progressive accumulation of genetic changes. The marginal zone of the B-cell follicle represents a well-defined compartment of the B area. Marginal zone-like B cells
“home” to an area outside the follicles of peripheral lymphoid tissues, such as the MALT tissues of the thyroid. These areas acquire organized lymphoid tissue as a result of chronic antigenic stimulation of lymphocytic thyroiditis. Its cellular composition is distinct from that of the follicle center while also distinct functionally in the immune response. Immunoglobulin (Ig) antigen receptor stimulation is thought to play an important role in clonal expansion of MALT lymphoma.
54 Ig heavy and light chain variable genes (VH and VL) expressed by MALT lymphoma show numerous point mutations in both VH and VL genes that are different relative to germ line genes. Furthermore, there is intraclonal sequence heterogeneity, indicative of ongoing somatic hypermutation. As Ig gene hypermutation is thought to occur at the post-GC stage of B-cell development, these findings suggest that the MALT lymphoma cell of origin is from post-GC, marginal zone B cells.
5,
54,
55,
56,
57,
58,
59 It is important to note, however, that gene rearrangements for Ig VH and VL and for T-cell receptor
β-chain genes are detected in lymphocytic thyroiditis, but to a much lesser degree.
32,
60,
61,
62 Therefore, PCR detection of a rearrangement cannot be used for diagnosis without confirmation by immunohistochemistry and histology. There are sequence similarities between the clonal bands of cells from lymphocytic thyroiditis and cells from the subsequent lymphoma.
34 Interestingly, different families of VH genes are detected in different lymphoma types: DLBCL shows VH3, whereas MALT lymphoma shows VH4 and VH3.
63 With transformation into a DLBCL, peripheral B cells of either GC or post-GC origin may be the cell of origin.
CLINICAL PRESENTATION
Primary thyroid gland lymphomas are estimated to represent up to 5% of all thyroid gland malignant neoplasms.
3,
4,
5,
10,
37 Lymphomas occur predominantly in middle to older aged women (mean age,
60 to 65 years), although a wide age range at initial presentation is reported (14 to 90 years).
2,
3,
4,
7,
9,
37,
38,
63,
64,
65,
66,
67,
68 Lymphomas occur chiefly in women, with a female to male ratio of 3 to 7:1.
2,
3,
4,
7,
9,
37,
38,
63,
64,
65,
66,
67,
68 Patients present with a mass lesion or goiter (overall enlargement), often with recent enlargement (sometimes rapidly). The mass results in additional obstructive symptoms related to compression. Additional symptoms include pain, dyspnea, dysphagia, hoarseness, choking, coughing, and hemoptysis.
3,
4,
9,
10,
38,
64,
69 Symptoms are usually experienced for a limited time (mean, 6 months duration), but MALT lymphoma tends to be a chronic, long-term clinical disorder. Most patients do not have B symptoms (fever, profound night sweats, weight loss, and anorexia), but they may develop in patients with DLBCL or other high-grade lymphomas. Antithyroid serum antibodies are identified in most patients, a finding correlated with the histologic presence of chronic lymphocytic (Hashimoto) thyroiditis.
3,
9,
10 Many patients are euthyroid, but hypothyroidism is common; very rarely patients will have hyperthyroidism.
4,
24,
38,
69,
70Radiographic iodine uptake studies usually show a “cold” or “cool” nodule but can show diffuse areas of low uptake,
3,
4,
10,
65,
71 although
mTcpertechnetate scintigraphy may show a “warm” nodule.
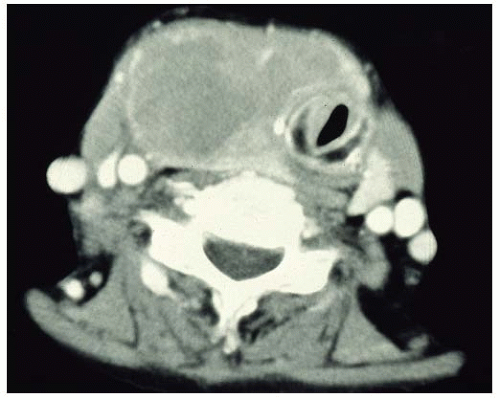
72 CT shows heterogeneous mass, sometimes with cystic change, whereas ultrasonographic features of primary thyroid lymphoma usually demonstrate a marked hypoechoic, asymmetrical pseudocystic mass compared with the residual thyroid tissue (
Fig. 17.1).
38,
73,
74 By
18F-Fluoro-deoxy-glucose positron emission tomography (FDG-PET), most lymphoma subtypes have high
18F-FDG avidity with the exception of MALT lymphoma and small lymphocytic lymphoma.
75 However,
18F-FDG PET may be more sensitive in MALT lymphoma with plasmacytic differentiation.
76 Incidental thyroid uptake with
18F-FDG-PET is not uncommon although it is typically diffuse, and focal lesions require further workup.
77 In some cases, no significant radiographic abnormality is noted.