Pituitary Gland Disorders
KEY CONCEPTS
![]() Pharmacologic therapy for acromegaly should be considered when surgery and irradiation are contraindicated, when there is poor likelihood of surgical success, when rapid control of symptoms is needed, or when other treatments have failed to normalize growth hormone (GH) and insulin-like growth factor-1 (IGF-1) concentrations.
Pharmacologic therapy for acromegaly should be considered when surgery and irradiation are contraindicated, when there is poor likelihood of surgical success, when rapid control of symptoms is needed, or when other treatments have failed to normalize growth hormone (GH) and insulin-like growth factor-1 (IGF-1) concentrations.
![]() Pharmacotherapy for acromegaly using dopamine agonists provides advantages of oral dosing and reduced cost compared to somatostatin analogs and pegvisomant. However, dopamine agonists effectively normalize IGF-1 concentrations in only 10% of patients. Therefore, somatostatin analogs remain the mainstay of therapy.
Pharmacotherapy for acromegaly using dopamine agonists provides advantages of oral dosing and reduced cost compared to somatostatin analogs and pegvisomant. However, dopamine agonists effectively normalize IGF-1 concentrations in only 10% of patients. Therefore, somatostatin analogs remain the mainstay of therapy.
![]() Blood glucose concentrations should be monitored frequently in the early stages of somatostatin analog therapy in all acromegalic patients.
Blood glucose concentrations should be monitored frequently in the early stages of somatostatin analog therapy in all acromegalic patients.
![]() Pegvisomant appears to be the most effective agent for normalizing IGF-1 concentrations. However, further study is needed to determine the long-term safety and efficacy of this agent for the treatment of acromegaly.
Pegvisomant appears to be the most effective agent for normalizing IGF-1 concentrations. However, further study is needed to determine the long-term safety and efficacy of this agent for the treatment of acromegaly.
![]() Recombinant GH is currently considered the mainstay of therapy for treatment of children with growth hormone-deficient (GHD) short stature. Prompt diagnosis of GHD and initiation of replacement therapy with recombinant GH is crucial for optimizing final adult heights.
Recombinant GH is currently considered the mainstay of therapy for treatment of children with growth hormone-deficient (GHD) short stature. Prompt diagnosis of GHD and initiation of replacement therapy with recombinant GH is crucial for optimizing final adult heights.
![]() All GH products are generally considered to be equally efficacious. The recommended dose for treatment of GHD short stature in children is 0.3 mg/kg/wk.
All GH products are generally considered to be equally efficacious. The recommended dose for treatment of GHD short stature in children is 0.3 mg/kg/wk.
![]() Pharmacologic agents that antagonize dopamine or increase the release of prolactin can induce hyperprolactinemia. Discontinuation of the offending medication and initiation of an appropriate therapeutic alternative usually normalize serum prolactin concentrations.
Pharmacologic agents that antagonize dopamine or increase the release of prolactin can induce hyperprolactinemia. Discontinuation of the offending medication and initiation of an appropriate therapeutic alternative usually normalize serum prolactin concentrations.
![]() Cabergoline appears to be more effective than bromocriptine for the medical management of prolactinomas and offers the advantage of less-frequent dosing and fewer adverse effects.
Cabergoline appears to be more effective than bromocriptine for the medical management of prolactinomas and offers the advantage of less-frequent dosing and fewer adverse effects.
![]() Although preliminary data do not suggest cabergoline has significant teratogenic potential, cabergoline is not recommended for use during pregnancy, and patients receiving cabergoline who plan to become pregnant should discontinue the medication as soon as pregnancy is detected.
Although preliminary data do not suggest cabergoline has significant teratogenic potential, cabergoline is not recommended for use during pregnancy, and patients receiving cabergoline who plan to become pregnant should discontinue the medication as soon as pregnancy is detected.
![]() Pharmacologic treatment of panhypopituitarism consists of glucocorticoids, thyroid hormone preparations, sex steroids, and recombinant GH, where appropriate, as lifelong replacement therapy.
Pharmacologic treatment of panhypopituitarism consists of glucocorticoids, thyroid hormone preparations, sex steroids, and recombinant GH, where appropriate, as lifelong replacement therapy.
INTRODUCTION
In the 1950s, Geoffrey Harris and his colleagues uncovered the physiologic importance of pituitary hormones and proposed the theory of neurohormonal regulation of the pituitary by the hypothalamus.1 Today the pituitary gland is recognized for its essential role in body homeostasis, and for this reason it is often referred to as the “master gland.” The hypothalamus and the pituitary gland are closely connected, and together they provide a means of communication between the brain and many of the body’s endocrine organs. The hypothalamus uses nervous input and metabolic signals from the body to control the secretion of pituitary hormones that regulate growth, thyroid function, adrenal activity, reproduction, lactation, and fluid balance.
ANATOMY AND PHYSIOLOGY
The hypothalamus (Fig. 60-1) is a small region at the base of the brain that receives autonomic nervous input from different areas of the body to regulate limbic functions, food and water intake, body temperature, cardiovascular function, respiratory function, and diurnal rhythms. In addition, the hypothalamus controls the release of hormones from the anterior and posterior regions of the pituitary gland. Neurons in the hypothalamus produce vasopressin and oxytocin and make many hormone-releasing factors that stimulate or inhibit the release of trophic hormones. At the base of the hypothalamus, a projection known as the median eminence is rich with nerve axons and blood vessels and provides both chemical and physical connections between the hypothalamus and the pituitary gland.
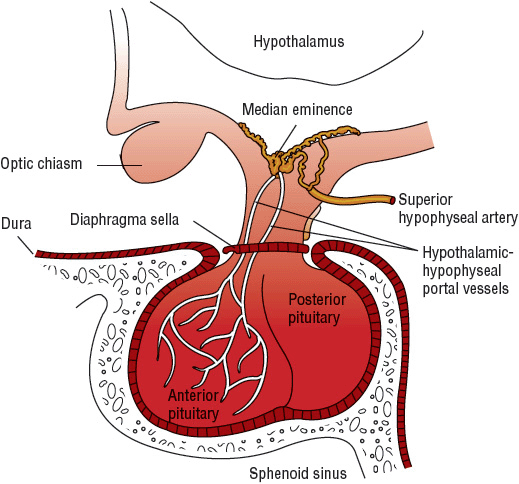
FIGURE 60-1 Pituitary gland.
The pituitary gland, also referred to as the hypophysis, is located at the base of the brain in a cavity of the sphenoid bone known as the sellaturcica. The pituitary is separated from the brain by an extension of the dura mater known as the diaphragma sellae. The pituitary is a very small gland, weighing between 0.4 and 1 g in adults. It is divided into two distinct regions: the anterior lobe, or adenohypophysis; and the posterior lobe, or the neurohypopituitary gland.physis (see Fig. 60-1).
The posterior pituitary gland secretes two major hormones: oxytocin and vasopressin (antidiuretic hormone) (Table 60-1). Oxytocin release from the posterior pituitary causes contraction of the smooth muscles in the breast during lactation. It also plays a role in uterine contraction during parturition. Vasopressin is essential for proper fluid balance and acts on the renal collecting ducts to conserve water. Oxytocin and vasopressin are synthesized in the paraventricular and supraoptic nuclei of the hypothalamus. The posterior pituitary gland contains the terminal nerve endings of these two nuclei as well as specialized secretory granules that release hormones in response to appropriate signals. Loss of anterior pituitary function does not necessarily affect the release of vasopressin or oxytocin because these hormones actually are synthesized in the hypothalamus.
TABLE 60-1 Pituitary Hormones
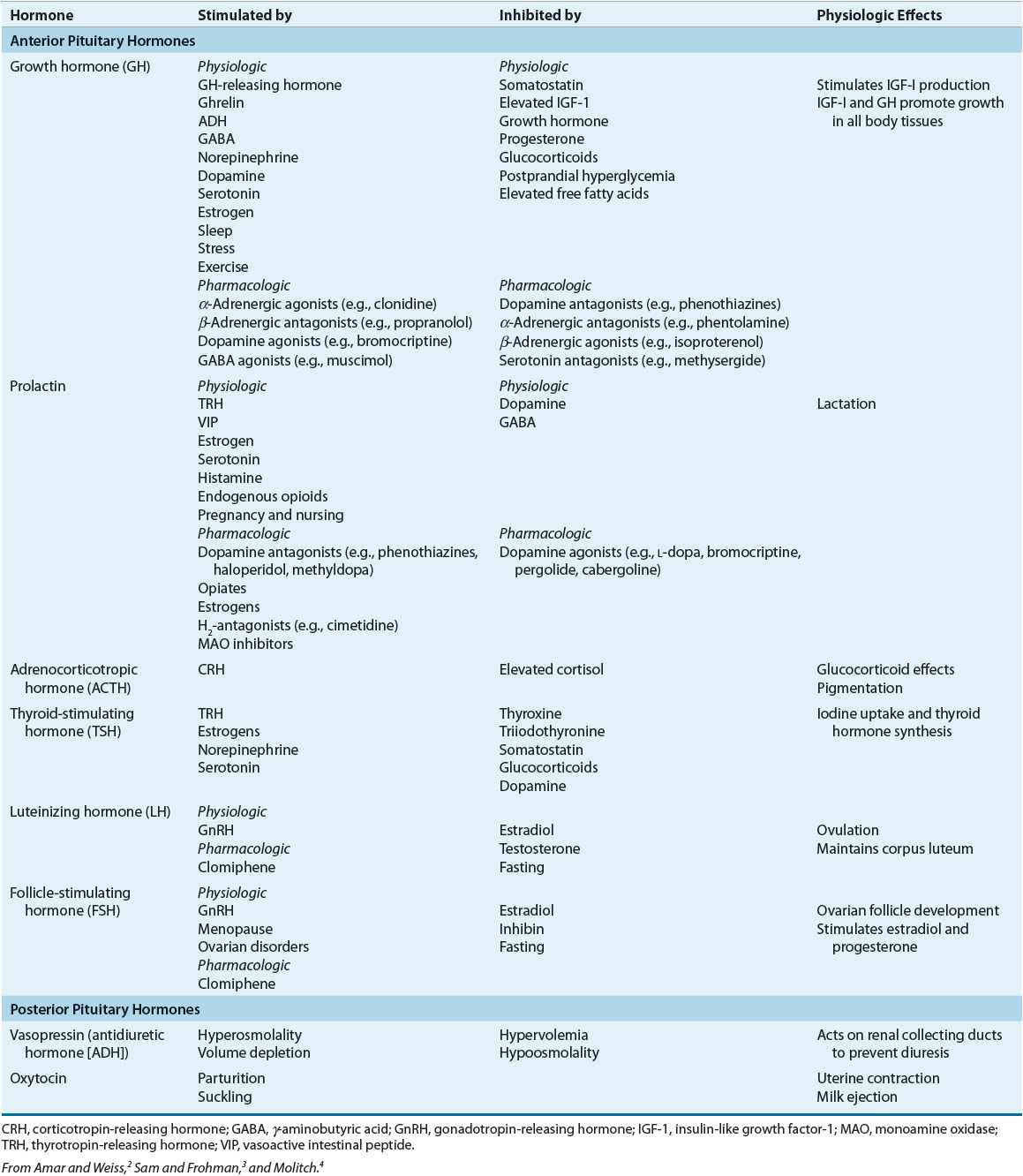
Unlike the posterior pituitary, the release of anterior pituitary hormones is not regulated by direct nervous stimulation but rather is controlled by specific hypothalamic-releasing and inhibitory hormones. The median eminence of the hypothalamus contains a large number of capillaries that converge to form a network of veins known as the hypothalamic–hypophyseal portal circulation. Inhibiting and releasing hormones synthesized in the neurons of the hypothalamus reach the anterior pituitary via the hypothalamic–hypophyseal portal vessels to control release of anterior pituitary hormones. Although there is a direct arterial blood supply to the anterior pituitary lobe, the hypothalamic–hypophyseal portal vessels provide the primary blood supply (see Fig. 60-1). In contrast to the posterior pituitary, the anterior pituitary lobe is extremely vascular and has the highest rate of blood flow of all body organs.
The specialized secretory cells of the anterior pituitary lobe secrete six major polypeptide hormones (see Table 60-1). These include growth hormone (GH) or somatotropin, adrenocorticotropic hormone (ACTH) or corticotropin, thyroid-stimulating hormone (TSH) or thyrotropin, prolactin, follicle-stimulating hormone (FSH), and luteinizing hormone (LH). The release of these hormones is regulated primarily by hypothalamic-releasing and inhibiting hormones. Thyrotropin-releasing hormone (TRH) stimulates anterior pituitary release of TSH and prolactin, corticotropin-releasing hormone (CRH) stimulates anterior pituitary release of ACTH, growth hormone-releasing hormone (GHRH) stimulates anterior pituitary release of GH, and gonadotropin-releasing hormone (GnRH) stimulates anterior pituitary release of LH and FSH. Hypothalamic release of somatostatin inhibits release of GH, and hypothalamic release of dopamine (prolactin inhibitory hormone) inhibits the secretion of prolactin. Prolactin differs from the other anterior lobe hormones in that an inhibiting factor, rather than a stimulating factor, is primarily responsible for controlling its secretion. In the absence of hypothalamic input, an excess of prolactin is produced, whereas a deficiency state of other anterior pituitary hormones results. Physiologic regulation and action of anterior and posterior pituitary hormones are summarized in Table 60-1.2–4
Destruction of the pituitary gland may result in secondary hypothyroidism, hypogonadism, adrenal insufficiency, GH deficiency, and hypoprolactinemia. The formation of certain types of pituitary tumors may result in pituitary hormone excess. Pituitary tumors may physically compress the pituitary and prevent the release of trophic hypothalamic factors that regulate pituitary hormones. In this chapter, the pathophysiology and role of pharmacotherapy in the treatment of acromegaly, short stature, hyperprolactinemia, and panhypopituitarism are discussed.
GROWTH HORMONE
GH has direct antiinsulin effects on lipid and carbohydrate metabolism. GH decreases utilization of glucose by peripheral tissues, increases lipolysis, and increases muscle mass. GH also stimulates gluconeogenesis in hepatocytes, impairs tissue glucose uptake, decreases insulin-receptor sensitivity, and impairs postreceptor insulin action. The growth-promoting effects of GH are largely mediated by insulin-like growth factors (IGFs) also known as somatomedins. GH stimulates the formation of IGF-1 in the liver as well as in other peripheral tissues. This anabolic peptide acts as a direct stimulator of cell proliferation and growth. There are two types of IGFs: IGF-1 and IGF-2. IGF-1 regulates growth to some extent before, and largely after, birth. In contrast, IGF-2 is thought to primarily regulate growth in utero.5 GH is secreted by the anterior pituitary in a pulsatile fashion, with several short bursts that occur mostly at night. Because of the short half-life of GH in the plasma (~30 minutes), measurements of circulating GH concentrations throughout the waking hours usually are very low or undetectable. Daytime GH pulses are most likely to occur after meals, following exercise, or during periods of stress. The greatest amount of GH secretion occurs during the night within the first 1 to 2 hours of slow-wave sleep (stage III or IV). Secretion of GH is lowest during infancy, increases slightly during childhood, reaches its peak during adolescence, and then begins to gradually decline during the middle-age years.3
Growth Hormone Excess
Acromegaly is a pathologic condition characterized by excessive production of GH. This is a rare disorder that affects approximately 50 to 70 adults per million.6 Gigantism, which is even more rare than acromegaly, is the excess secretion of GH prior to epiphyseal closure in children.7 Patients diagnosed with acromegaly are reported to have a two- to threefold increase in mortality, usually related to cardiovascular, respiratory, or neoplastic disease.8–10 Most patients are middle-aged at the time of diagnosis, and this disorder does not appear to affect one sex to a greater extent than the other. The most common cause of excess GH secretion in acromegaly is a GH-secreting pituitary adenoma, accounting for over 90% of all cases.8 Rarely, acromegaly is caused by ectopic GH-secreting adenomas, GH cell hyperplasia, or excess GHRH secretion, or is one of the manifestations of multiple endocrine neoplasia syndrome type 1, McCune–Albright’s syndrome, or the Carney complex, all very rare hypersecretory endocrinopathies.8
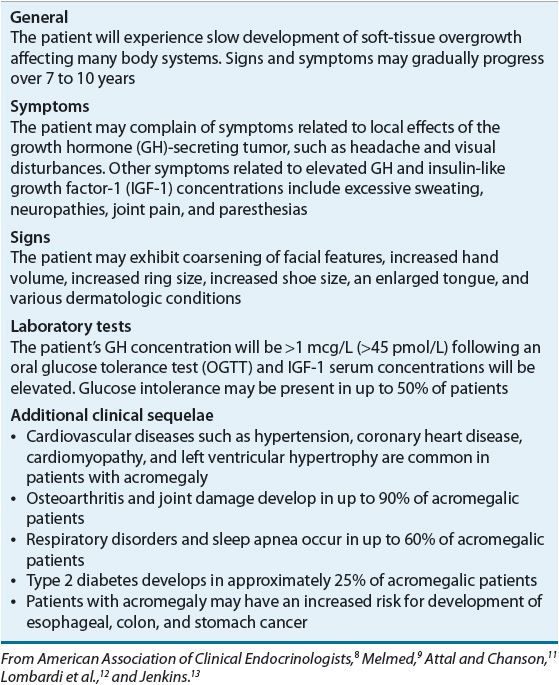
The clinical signs and symptoms of acromegaly develop gradually over an extended period of time. In fact, because of the subtle and slowly developing changes in physical appearance caused by GH excess, most patients are not definitively diagnosed with acromegaly until 7 to 10 years after the presumed onset of excessive GH secretion.9 Excessive secretion of GH and IGF-1 adversely affects several organ systems. Almost all acromegalic patients will present with physical signs and symptoms of soft-tissue overgrowth. Table 60-2 summarizes the classic clinical presentation of patients with acromegaly.8–13 Some patients with acromegaly present with only a few of these classic signs and symptoms, making recognition of this disease extremely difficult.
TABLE 60-2 Clinical Presentation of Acromegaly
The diagnosis of acromegaly is based on a combination of diagnostic tests and clinical signs and symptoms. Random measures of plasma GH levels are not usually dependable because of the pulsatile pattern of release. However, some clinicians exclude diagnosis of acromegaly in the presence of a random GH <0.4 mcg/L (<18 pmol/L) and IGF-1 that is normal for age and sex.8 The oral glucose tolerance test (OGTT) is commonly used as an important diagnostic tool. Postprandial hyperglycemia inhibits the secretion of GH for at least 1 to 2 hours. Therefore, an oral glucose load would be expected to suppress GH concentrations. However, patients with acromegaly continue to secrete GH during the OGTT. Because GH stimulates the production of IGF-1, serum IGF-1 concentrations can also be measured to aid in the diagnosis of acromegaly. Circulating IGF-1 is cleared from the body at a much slower rate than is GH, and measurements can be collected at any time of the day to identify patients with GH excess.9 Current criteria for the diagnosis of acromegaly include failure of GH suppression <1 mcg/L (<45 pmol/L) following an OGTT in the presence of elevated IGF-1 serum concentrations.8,14 With the development of more sensitive GH and IGF-1 assays, the American Association of Clinical Endocrinologists (AACE) suggests lowering the cutoff for GH suppression to <0.4 mcg/L (<18 pmol/L). Insulin-like growth factor 1 binding protein 3 (IGFBP-3) also can be measured because it is positively regulated by GH and binds to circulating IGF-1 with high affinity. This test may prove useful in the future in monitoring response to therapy but, at present, AACE does not recommend IGFBP-3 measurement for the purpose of clinical management.8 Computed tomography and magnetic resonance imaging of the pituitary are important diagnostic tests to confirm the presence of a pituitary adenoma.8,14
TREATMENT
Acromegaly
The primary treatment goals for patients diagnosed with acromegaly are to reduce GH and IGF-1 concentrations, improve the clinical signs and symptoms of the disease, and decrease mortality.8,15–17 Many clinicians define biochemical control of acromegaly as suppression of GH concentrations to <1 mcg/L (<45 pmol/L) after a standard OGTT in the presence of normal IGF-1 serum concentrations, although some argue for a lower cutoff GH value of 0.4 mcg/L (18 pmol/L) due to the availability of more sensitive test methods.16 The treatment of choice for most patients with acromegaly is transsphenoidal surgical resection of the GH-secreting adenoma.9,15,16 Postsurgical cure rates have been reported to range from 50% to 90%, depending on the type of adenoma and the expertise of the neurosurgeon.8,16,17 Complications of transsphenoidal surgery are relatively infrequent and include cerebrospinal fluid leak, meningitis, arachnoiditis, diabetes insipidus, and pituitary failure.8 For patients who are poor surgical candidates, those who have not responded to surgical or medical interventions, or others who refuse surgical or medical treatment, radiation therapy may be considered. Radiation, however, may require several years to relieve the symptoms of acromegaly.
Because neither radiation therapy nor surgery will cure all patients with acromegaly, adjuvant drug therapy is often needed to control symptoms.9,17,18
Pharmacologic Therapy
![]() Drug therapy should be considered as primary therapy for acromegalic patients who prefer medical therapy, are poor surgical candidates, or when there is a poor likelihood of surgical success. Drug therapy should be considered as adjunctive therapy in the presence of persistent disease after surgery.8 Pharmacologic treatment options include dopamine agonists, somatostatin analogs, and the GH receptor antagonist pegvisomant. Dopamine agonists such as bromocriptine and cabergoline are effective in a small subset of patients and provide the advantages of oral dosing and reduced cost. Somatostatin analogs are more effective than dopamine agonists, reducing GH concentrations and normalizing IGF-1 in approximately 50% to 60% of patients. Pegvisomant, a GH receptor antagonist, is highly effective in normalizing IGF-1 concentrations in up to 97% of patients in the first year and in 60% over 5 years.
Drug therapy should be considered as primary therapy for acromegalic patients who prefer medical therapy, are poor surgical candidates, or when there is a poor likelihood of surgical success. Drug therapy should be considered as adjunctive therapy in the presence of persistent disease after surgery.8 Pharmacologic treatment options include dopamine agonists, somatostatin analogs, and the GH receptor antagonist pegvisomant. Dopamine agonists such as bromocriptine and cabergoline are effective in a small subset of patients and provide the advantages of oral dosing and reduced cost. Somatostatin analogs are more effective than dopamine agonists, reducing GH concentrations and normalizing IGF-1 in approximately 50% to 60% of patients. Pegvisomant, a GH receptor antagonist, is highly effective in normalizing IGF-1 concentrations in up to 97% of patients in the first year and in 60% over 5 years.
Dopamine Agonists
![]() In normal healthy adults, dopamine agonists cause an increase in GH production. However, when these agents are given to patients with acromegaly, there is a paradoxical decrease in GH production. Most clinical experience with the use of dopamine agonists in acromegaly is with bromocriptine or cabergoline. Other agents such as pergolide, quinagolide, and lisuride also have been used but are not available in the United States. Bromocriptine and cabergoline are semisynthetic ergot alkaloids that act as dopamine-receptor agonists. Most trials assessing the efficacy of bromocriptine in the treatment of acromegaly were conducted in the 1970s and early 1980s and determined that certain subsets of acromegalic patients with high circulating concentrations of prolactin have a favorable response to drug therapy with bromocriptine.19 A review evaluating 34 studies concluded that therapy with bromocriptine was effective in suppressing mean serum GH levels to <5 mcg/L (<225 pmol/L) in approximately 20% of patients.20 While only 10% of patients experience normalization of IGF-1 concentrations with bromocriptine therapy, greater than 50% of patients treated with bromocriptine experience improvement in symptoms of acromegaly.8,19 According to AACE guidelines, cabergoline appears used more commonly than bromocriptine. A recent meta-analysis of 15 studies concluded that cabergoline as monotherapy was effective in normalizing IGF-1 levels in 34% of patients and resulted normalization of IGF-1 levels in 52% of patients when added to a somatostatin analog in those unresponsive to somatostatin analog monotherapy.21
In normal healthy adults, dopamine agonists cause an increase in GH production. However, when these agents are given to patients with acromegaly, there is a paradoxical decrease in GH production. Most clinical experience with the use of dopamine agonists in acromegaly is with bromocriptine or cabergoline. Other agents such as pergolide, quinagolide, and lisuride also have been used but are not available in the United States. Bromocriptine and cabergoline are semisynthetic ergot alkaloids that act as dopamine-receptor agonists. Most trials assessing the efficacy of bromocriptine in the treatment of acromegaly were conducted in the 1970s and early 1980s and determined that certain subsets of acromegalic patients with high circulating concentrations of prolactin have a favorable response to drug therapy with bromocriptine.19 A review evaluating 34 studies concluded that therapy with bromocriptine was effective in suppressing mean serum GH levels to <5 mcg/L (<225 pmol/L) in approximately 20% of patients.20 While only 10% of patients experience normalization of IGF-1 concentrations with bromocriptine therapy, greater than 50% of patients treated with bromocriptine experience improvement in symptoms of acromegaly.8,19 According to AACE guidelines, cabergoline appears used more commonly than bromocriptine. A recent meta-analysis of 15 studies concluded that cabergoline as monotherapy was effective in normalizing IGF-1 levels in 34% of patients and resulted normalization of IGF-1 levels in 52% of patients when added to a somatostatin analog in those unresponsive to somatostatin analog monotherapy.21
In the United States, bromocriptine is commercially available as 0.8 and 2.5-mg oral tablets and 5-mg oral capsules. The 0.8-mg tablet is indicated as adjunctive therapy in type 2 diabetes mellitus. In acromegalic patients, significant reductions in GH concentrations are observed within 1 to 2 hours of oral dosing. This effect persists for at least 4 to 5 hours. An overall clinical response in acromegalic patients typically occurs after 4 to 8 weeks of continuous bromocriptine therapy. For treatment of acromegaly, bromocriptine is initiated at a dose of 1.25 mg (1/2 of a 2.5-mg tablet) at bedtime and is increased by 1.25-mg increments every 3 to 4 days as needed. Doses as high as 86 mg/day have been used for treatment of acromegaly, but clinical studies have shown that dosages >20 or 30 mg daily do not offer additional benefits in the suppression of GH. When used for treatment of acromegaly, the duration of action of bromocriptine is shorter than that for treatment of hyperprolactinemia. Therefore, the total daily dose of bromocriptine should be divided into three or four doses.
Cabergoline is commercially available as 0.5 mg tablets. Use in acromegaly is considered off-label, and dosing is typically initiated at 0.5 mg twice weekly and increased as needed to 0.5 mg every other day. Doses up to 7 mg/wk (0.5 mg twice daily) have been reported in trials.
The most common adverse effects of dopamine agonist therapy include CNS symptoms such as headache, lightheadedness, dizziness, nervousness, and fatigue. GI effects such as nausea, abdominal pain, or diarrhea also are very common. Some patients may need to take dopamine agonists with food to decrease the incidence of adverse GI effects. Most adverse effects are seen early in the course of therapy and tend to decrease with continued treatment.8,19 Dopamine agonists may cause thickening of bronchial secretions and nasal congestion. Rare cases of psychiatric disturbances, pleural diseases, and an erythromelalgic syndrome (painful paroxysmal dilation of the blood vessels in the skin of the feet and lower extremities) have been reported with dopamine agonist use. These conditions appear to be associated with higher doses and prolonged duration of therapy.8,19
Dopamine agonists are not FDA-approved for use during pregnancy. However, surveillance of women who took dopamine agonists throughout pregnancy does not suggest that dopamine agonists are associated with an increased risk for birth defects.22 If a woman becomes pregnant while taking dopamine agonists, the risks and benefits of therapy should be fully considered. In most cases, the benefits of successful therapy outweigh the risks, and dopamine agonist therapy should be continued if it is effective in improving symptoms and reducing elevated GH concentrations.
Other dopamine agonists that have been used to treat acromegaly include pergolide, lisuride, and quinagolide. Pergolide is no longer commercially available, and lisuride and quinagolide are not commercially available in the United States. Because of the potential cost advantages and convenience of oral administration, dopamine agonists are often considered for treatment of acromegaly prior to initiation of somatostatin analogs. However, the availability of long-acting somatostatin analogs has made these agents more attractive for first-line treatment of acromegaly.
Somatostatin Analogs
Octreotide and lanreotide are long-acting somatostatin analogs that are more potent in inhibiting GH secretion than endogenous somatostatin.23,24 These agents also suppress the LH response to GnRH; decrease splanchnic blood flow; and inhibit secretion of insulin, vasoactive intestinal peptide (VIP), gastrin, secretin, motilin, serotonin, and pancreatic polypeptide. Pasireotide is a somatostatin analog that has a broader affinity for somatostatin receptor subtypes than octreotide or lanreotide. The binding to additional subtypes of somatostatin receptors may result in greater GH inhibition compared to octreotide or lanreotide and efficacy of pasireotide in the presence of octreotide or lanreotide-resistant adenomas.23
Octreotide (Sandostatin) injection is commercially available in the United States for subcutaneous or IV administration. A long-acting intramuscular formulation of octreotide (Sandostatin LAR) is available for monthly administration. In addition to the treatment of acromegaly, octreotide has many other therapeutic uses, including the treatment of carcinoid tumors, vasoactive intestinal peptide-secreting tumors (VIPomas), GI fistulas, variceal bleeding, diarrheal states, and irritable bowel syndrome.
The efficacy of octreotide for treatment of acromegaly has been determined by two major multicenter trials.25,26 These studies determined that drug therapy with octreotide suppresses mean serum GH concentrations to <5 mcg/L (<225 pmol/L) and normalizes serum IGF-1 concentrations in 50% to 60% of acromegalic patients. Octreotide also is beneficial in reducing the clinical signs and symptoms of acromegaly. In a 6-month multicenter trial, 70% of patients experienced significant relief of headaches.26 In some patients, relief of headache symptoms occurred within minutes of octreotide administration. In addition, middle-finger circumference was reduced significantly, and 50% to 75% of patients experienced improvement in symptoms of excessive perspiration, fatigue, joint pain, and cystic acne. Long-term follow-up of patients treated with octreotide LAR for up to 9 years showed octreotide therapy to be safe and effective for long-term use in acromegalic patients.27 Octreotide also has been shown to improve the cardiovascular manifestations of acromegaly and to halt pituitary tumor growth, with some patients experiencing tumor regression.25–28 Data from more recent studies indicate that shrinkage of pituitary tumor mass during octreotide therapy occurs in approximately 50% of patients.29
The pharmacodynamic effects of long-acting octreotide are similar to those of subcutaneously administered octreotide. Single monthly doses of long-acting octreotide have been shown to be at least as effective as daily doses of subcutaneous octreotide administered in divided doses three times daily in normalizing IGF-1 levels and maintaining suppression of mean serum GH concentrations.30 Trials evaluating the efficacy of long-acting octreotide in acromegalic patients who previously had responded to subcutaneously administered octreotide have reported sustained suppression of GH concentrations to <5 mcg/L (<225 pmol/L) and normalization of IGF-1 in patients following 1 year of therapy.31
Response to long-term therapy with octreotide is related to the presence and increased quantity of functioning somatostatin receptors located in the pituitary adenoma. Identification of patients who most likely will respond to octreotide, prior to initiation of therapy, is important when considering the high cost of this medication and the inconvenience of subcutaneous or intramuscular drug administration. Suppression of serum GH concentrations after a single 50-mcg dose of octreotide has been used to predict a favorable long-term response to octreotide therapy.32,33
The initial dose of octreotide for treatment of acromegaly is usually 100 mcg administered three times daily followed by either titration to a maximum of 1500 mcg/day or transition to long-acting octreotide.8,23,24 Some clinicians recommend a starting dose of 50 mcg every 8 hours, then increasing the dose to 100 mcg every 8 hours after 1 week, to improve the patient’s tolerance of adverse GI effects. The dose can be increased by increments of 50 mcg every 1 to 2 weeks based on mean serum GH and IGF-1 concentrations. Patients who experience a significant rise in GH prior to the end of the 8-hour dosing interval may benefit from decreasing the dosing interval to every 4 to 6 hours. Although doses as high as 1,500 mcg/day have been used, doses >600 mcg daily generally do not offer additional benefits, and most patients are adequately managed with 100 to 200 mcg three times daily.8,24 Patients who have been maintained on subcutaneous octreotide for at least 2 weeks and have shown response to therapy can be converted to the long-acting depot form of octreotide. The initial dose of long-acting octreotide is 20 mg administered intramuscularly in the gluteal region every 28 days. Steady-state serum concentrations are not obtained until after 3 months of therapy. Therefore, dosage adjustments for long-acting octreotide should not be considered until after this time. Some patients may require additional subcutaneous injections during the initial dose-titration phase in order to control symptoms. In patients who achieve >50% reduction in GH levels to 30 mg every 4 weeks, some may have added response to a higher-dose regimen of 60 mg every 4 weeks.34
A long-acting, intramuscular formulation of lanreotide (lanreotide LA) for twice monthly administration has been available in Europe for many years. In 2007, a new formulation of lanreotide (Somatuline Depot) was approved for use in the United States for monthly deep subcutaneous administration. The efficacy of this preparation of lanreotide for the treatment of acromegaly has been evaluated in several prospective multicenter clinical trials involving treatment-experienced patients who were switched from intramuscular octreotide LAR or intramuscular lanreotide LA to monthly deep subcutaneous lanreotide.35,36 These studies have determined that deep subcutaneous lanreotide suppresses mean serum GH concentrations to <5 mcg/L (<225 pmol/L) and normalizes serum IGF-1 concentrations in acromegalic patients to a similar extent as octreotide LAR and lanreotide LA. A 4-year follow-up of 23 patients treated with monthly deep subcutaneous lanreotide reported the drug to be well tolerated during long-term therapy with mean serum GH concentrations <5 mcg/L (225 pmol/L) in 62% of patients and normalization of serum IGF-1 concentrations in 43% of patients.37 Analyses of trials investigating the effects of lanreotide on pituitary tumor mass have shown shrinkage in 66% to 77% of patients.35 Well-designed trials directly comparing the efficacy of intramuscular octreotide LAR to deep subcutaneous lanreotide are currently lacking. However, these two agents are generally regarded to have comparable efficacy.8
Lanreotide (Somatuline Depot) is commercially available in the United States as 60-, 90-, and 120-mg prefilled syringes. In contrast to octreotide LAR, lanreotide injection does not need to be reconstituted prior to administration. The initial recommended dose of lanreotide is 90 mg given by deep subcutaneous injection in the superior external quadrant of the buttock every 28 days. Injection sites should be alternated between the left and right side. The initial dose should be reduced to 60 mg every 28 days for patients with moderate or severe renal or hepatic impairment. After 3 months of therapy, the dose may then be titrated based on serum GH concentrations, serum IGF-1 concentrations, and control of clinical signs and symptoms of acromegaly.36 Long-acting deep subcutaneous lanreotide injection in doses >120 mg every 28 days has not been studied.
The most common adverse effects of somatostatin analog therapy are GI disturbances such as diarrhea, nausea, abdominal cramps, malabsorption of fat, and flatulence.16,23 GI adverse effects occur in approximately 75% of patients but usually subside within 10 to 14 days of continued treatment.23 Octreotide has been reported to cause injection-site pain (4% to 31%), conduction abnormalities and arrhythmias (9%), subclinical hypothyroidism (2% to 12%), biliary tract disorders (4% to 50%), and abnormalities in glucose metabolism (2% to 18%).20,23 Lanreotide has been reported to cause injection-site reactions (9%), sinus bradycardia (3%), hypertension (5%), biliary tract disorders (20%), and abnormalities in glucose metabolism (7%).36
Somatostatin analogs also inhibit cholecystokinin release and gallbladder motility, predisposing patients to the development of cholelithiasis.23 The development of gallstones is a long-term adverse effect of somatostatin analog therapy and is largely dependent on geographic factors, dietary habits, and length of therapy.20,24 The incidence of gallstones in acromegalic patients receiving octreotide and lanreotide increases with length of therapy and has been reported to range from 20% to 50%.24–26 However, most patients are asymptomatic, and the diagnosis of cholelithiasis usually is made following an ultrasonographic study that is not prompted by patient symptoms. It has been estimated that only 1% of patients will develop symptomatic gallstones during 1 year of octreotide treatment.24 Because somatostatin analog-induced gallstones usually are present without clinical symptoms, prophylactic cholecystectomy or medical therapy with ursodeoxycholic acid for acromegalic patients with asymptomatic gallstones usually is not recommended.9,16 A small number of studies have suggested that the incidence of gallstone development may be lower with long-acting octreotide compared to subcutaneous octreotide.30,31 However, further studies are needed to confirm this observation.
![]() The effect of somatostatin analogs on glucose metabolism in patients with acromegaly is multifactorial. Decreases in serum GH concentrations induced by somatostatin analogs should result in decreased hepatic gluconeogenesis and increased insulin-receptor sensitivity. However, somatostatin analogs also decrease insulin secretion and increase IGFBP-1, which is known to inhibit the insulin-like effects of IGF-1. In addition, somatostatin analogs delay the GI absorption of glucose, which may further alter glucose metabolism in acromegalic patients.38 Small studies conducted in acromegalic patients receiving octreotide have reported improvement in insulin sensitivity as well as impaired insulin secretion.39 Risk factors associated with worsening glucose tolerance included female sex and elevated baseline insulin values. Although somatostatin analogs appear to have a beneficial effect on glucose tolerance in most patients, glucose determinations should be obtained frequently in the early stages of therapy in all acromegalic patients.
The effect of somatostatin analogs on glucose metabolism in patients with acromegaly is multifactorial. Decreases in serum GH concentrations induced by somatostatin analogs should result in decreased hepatic gluconeogenesis and increased insulin-receptor sensitivity. However, somatostatin analogs also decrease insulin secretion and increase IGFBP-1, which is known to inhibit the insulin-like effects of IGF-1. In addition, somatostatin analogs delay the GI absorption of glucose, which may further alter glucose metabolism in acromegalic patients.38 Small studies conducted in acromegalic patients receiving octreotide have reported improvement in insulin sensitivity as well as impaired insulin secretion.39 Risk factors associated with worsening glucose tolerance included female sex and elevated baseline insulin values. Although somatostatin analogs appear to have a beneficial effect on glucose tolerance in most patients, glucose determinations should be obtained frequently in the early stages of therapy in all acromegalic patients.
Growth Hormone Receptor Antagonist
![]() Pegvisomant (Somavert) is a genetically engineered GH derivative that binds to, but does not activate, GH receptors and inhibits IGF-1 production. This agent is different from other medications used in the management of acromegaly because it does not inhibit GH production; rather, it blocks the physiologic effects of GH on target tissues. Therefore, GH concentrations remain elevated during therapy, and response to treatment is evidenced by a reduction in IGF-1 concentrations. Unlike somatostatin analogs, the pharmacologic activity of pegvisomant does not depend on the presence and quantity of somatostatin receptors in the pituitary tumor.40 Studies evaluating the clinical efficacy of pegvisomant in acromegalic patients have reported a dose-dependent normalization of IGF-1 concentrations in 54% to 89% of patients after 12 weeks of therapy and in 97% of patients after 1 year of therapy.40,41 Significant improvements in the clinical signs and symptoms of acromegaly were reported and persisted throughout the 1-year treatment period.41 An ongoing, international post-marketing surveillance registry (ACROSTUDY) has recently reported 5-year data in patients treated with pegvisomant. In patients who predominantly had failed prior medical or surgical therapy, IGF-1 normalized in 63%. Investigators note that failure to maintain IGF-1 normalization may reflect suboptimal dosing strategies or more advanced disease than reported in the original studies.42
Pegvisomant (Somavert) is a genetically engineered GH derivative that binds to, but does not activate, GH receptors and inhibits IGF-1 production. This agent is different from other medications used in the management of acromegaly because it does not inhibit GH production; rather, it blocks the physiologic effects of GH on target tissues. Therefore, GH concentrations remain elevated during therapy, and response to treatment is evidenced by a reduction in IGF-1 concentrations. Unlike somatostatin analogs, the pharmacologic activity of pegvisomant does not depend on the presence and quantity of somatostatin receptors in the pituitary tumor.40 Studies evaluating the clinical efficacy of pegvisomant in acromegalic patients have reported a dose-dependent normalization of IGF-1 concentrations in 54% to 89% of patients after 12 weeks of therapy and in 97% of patients after 1 year of therapy.40,41 Significant improvements in the clinical signs and symptoms of acromegaly were reported and persisted throughout the 1-year treatment period.41 An ongoing, international post-marketing surveillance registry (ACROSTUDY) has recently reported 5-year data in patients treated with pegvisomant. In patients who predominantly had failed prior medical or surgical therapy, IGF-1 normalized in 63%. Investigators note that failure to maintain IGF-1 normalization may reflect suboptimal dosing strategies or more advanced disease than reported in the original studies.42
Adverse effects include injection-site pain, GI complaints such as nausea and diarrhea, and flu-like symptoms. Significant elevations in hepatic aminotransferase concentrations, which are generally reversible upon discontinuation of the drug, have been reported in approximately 25% of patients.43 As a result, hepatic function tests should be monitored very closely during therapy as outlined in the product labeling, and the drug should be used with caution in patients with baseline elevations in hepatic aminotransferase concentrations. GH concentrations may increase significantly during the first 6 months of therapy. Tumor growth has been reported in a small number of patients and there are theoretical concerns that the lack of GH feedback regulation on tumors that lead to persistently elevated GH concentrations may stimulate tumor growth or result in other long-term adverse effects. Interim results of the ongoing ACROSTUDY suggest that the rate of tumor growth of 3.2% is comparable to the background rate in acromegaly, and the incidence of hepatic aminotransferases greater than three times upper limit of normal is low (2.5%).43
Pegvisomant is commercially available in the United States for daily subcutaneous use. The first dose should be administered under the supervision of a physician as a 40-mg loading dose. Subsequent doses are self-administered by the patient starting at a dose of 10 mg daily. The dose can be adjusted in 5-mg increments based on serum IGF-1 concentrations every 4 to 6 weeks.43
Based on the available data, pegvisomant appears to be among the most effective agents for normalizing IGF-1 serum concentrations. Current guidelines for acromegaly management suggest pegvisomant therapy for patients who have failed to achieve normalization of IGF-1 serum concentrations with other treatments.8
Combination Therapy
Several small studies have suggested that combination therapy with somatostatin analogs, dopamine agonists, or pegvisomant may be more beneficial than monotherapy with either drug alone.8,21,44 Several of these trials have used doses lower than those typically used for monotherapy in order to try to minimize the risk of additive adverse effects. Because of the potential for additive adverse effects, combination therapy should be considered as a therapeutic option only for refractory patients who have not fully responded to monotherapy.8
Personalized Pharmacotherapy
The genetics of growth hormone and receptors have been well-studied.45 At this time, the data are most abundant with pegvisomant. As pegvisomant acts at the growth hormone receptor, researchers have investigated response to GH receptor variants. In patients with exon 3-deleted GH receptors, lower doses and fewer months were needed to obtain IGF-1 normalization.46 However, recommendations regarding how therapy can be individualized to maximize patient benefit are not yet available.8



