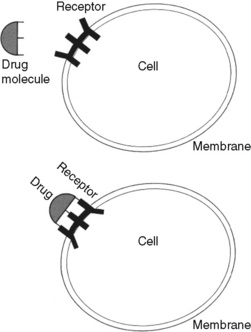
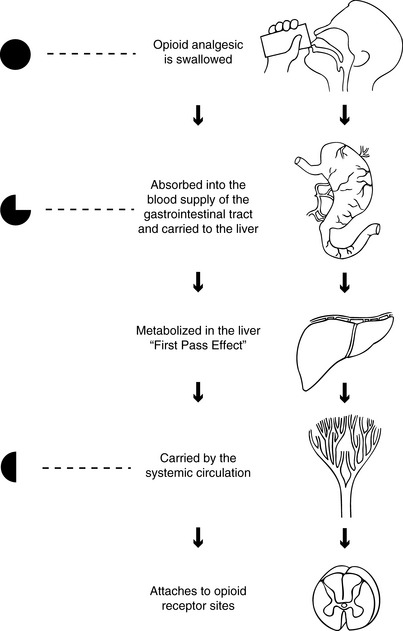
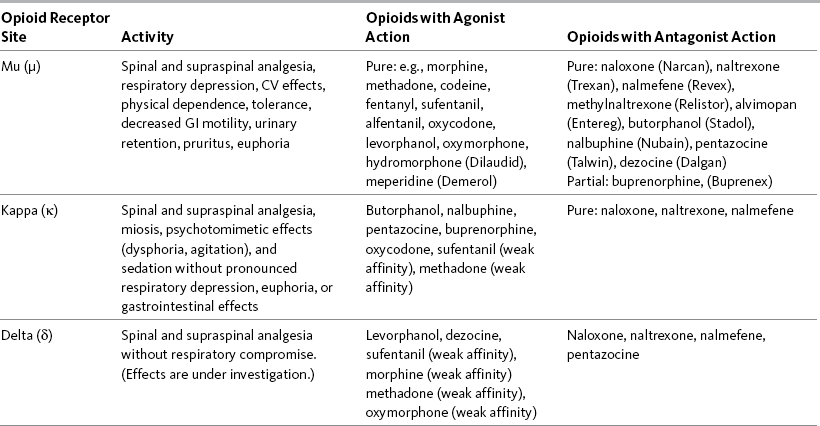
Chapter 11 THIS chapter expands on the underlying mechanisms of the opioid analgesics presented in Section I of this book. Several pharmacokinetic and pharmacodynamic concepts are explained. Terms such as opioid naïve and opioid tolerant are distinguished, and addiction, pseudoaddiction, physical dependence, tolerance, and cross tolerance are defined. The research and recommended diagnostic and treatment approaches for opioid-induced hyperalgesia are presented. To understand the appropriate use of opioids in the treatment of pain, it is valuable to review the mechanisms that are described by the term nociception. Nociception refers to the normal functioning of physiologic systems that lead to the perception of noxious stimuli as painful. In short, it means “normal” pain transmission. Nociception is described in Section I of this book. Review of Section I is advised so that the following discussion of the underlying mechanisms of the analgesia and adverse effects of opioids is clear. As explained in the discussion of nociception in Section I, the modulation (inhibition) of pain involves the release of dozens of neurochemicals by peripheral and central systems. Endogenous opioids (internal naturally-occurring), for example, are found throughout the periphery and central nervous system (CNS), including in the cardiovascular (CV) and gastrointestinal (GI) systems, pituitary gland, and in immune cells (Machelska, 2007; Mousa, 2003; Murphy, 2006; Rittner, Brack, 2007). It is thought that opioids given therapeutically activate endogenous pain-modulating systems and produce analgesia and other effects by binding to opioid receptor sites and mimicking the action of endogenous opioid compounds. Endogenous opioids are composed of three distinct families of peptides (naturally occurring compounds of two or more amino acids), all pharmacologically related to morphine: enkephalins, dynorphins, and β-endorphins (Gutstein, Akil, 2006; Inturrisi, 2002). Other endogenous peptides that have been more recently discovered—the endomorphins and orphanin-FQ—also appear to be important in pain processing, but their roles are yet poorly understood; the endomorphins bind selectively to the mu receptor, and orphanin-FQ is a ligand for another receptor, which is known as opioid receptor–like 1 (ORL1). Drugs exert their effects on the body by interacting with specialized macromolecular components in cells called drug receptors. Drug receptors usually are cellular proteins, but can be enzymes, carbohydrate residues, and lipids. The binding of drug molecules to their specific receptor molecules often is described as similar to a key fitting a lock (Figure 11-1). Binding affinity refers to the strength of attachment of a drug to the receptor site, and drugs bind with varying strength. The electromagnetic forces produced by the bond between a drug and receptor distort the configuration of the receptor molecule, changing its biochemical properties and functions. The body’s responses to the drug are a result of these changes (Bateman, Eddleston, 2007). Researchers think that receptors evolved for the purpose of interacting with endogenous compounds. The endogenous opioid system is an excellent example of this interaction. Opioid receptors are particularly abundant in the periacqueductal gray (PAG) and dorsal horn of the spinal cord. They are also located in the brainstem, thalamus, and cortex. Their presence in the midbrain PAG, nucleus raphe magnus, and the rostral ventral medulla help to inhibit pain via the descending modulatory system (Inturrisi, 2002). (See Section I and Figure I-2, D on pp. 4-5.) Opioids also reduce pain transmission by activating inhibitory pathways that originate segmentally (in the spinal cord) and supraspinally (Inturrisi, 2002). For example, the gamma aminobutyric acid (GABA) pathway is one of the major inhibitory neurotransmitter systems (see Section I), and opioids can activate the GABA system, leading to inhibition of pain transmission (Bridges, Thompson, Rice, 2001). When an opioid binds to the mu, delta, or kappa opioid receptor sites as an agonist, it produces analgesia as well as unwanted effects, such as nausea, constipation, and respiratory depression. Antagonists are drugs that also bind to opioid receptors but produce no analgesia. If an antagonist is present, it competes with opioid molecules for binding sites on the receptors. When a drug binds to any of the opioid receptor sites as an antagonist, analgesia and other effects are blocked. For example, naloxone, an opioid antagonist, can bind to the mu site and reverse analgesia and other opioid adverse effects, such as respiratory depression and sedation (Gutstein, Akil, 2006). See Table 11-1 for a summary of actions at opioid receptor type. Table 11-1 Summary of Actions at Opioid Receptor Type CV, Cardiovascular; GI, gastrointestinal. The effects (activity) a drug produces depends on the type(s) of opioid receptor(s) to which the drug binds and whether the drug acts as an agonist or an antagonist at that opioid receptor type. When a drug binds to any of these receptor sites as an agonist, it produces analgesia and other effects. When a drug binds to any of the opioid receptor sites as an antagonist, analgesia and other effects are blocked. Table 11-1 summarizes the activity of drugs when they bind to any of three opioid receptor types that are involved in analgesia. From Pasero, C., & McCaffery, M. Pain assessment and pharmacologic management, p. 285, St. Louis, Mosby. Data from Fine, P., & Portenoy, R. K. (2007). A clinical guide to opioid analgesia. New York, Vendome Group, LLC; Gutstein, H. B., & Akil, H. (2006). Opioid analgesics. In L. L. Brunton, J. S. Lazo, & K. L. Parker (Eds.), Goodman & Gilman’s the pharmacological basis of therapeutics, ed 11, New York, McGraw-Hill; Hanks, G., Cherny, N. I., & Fallon, M. Opioid analgesic therapy. In D. Doyle, G. Hanks, N. I. Cherny, et al. (Eds.), Oxford textbook of palliative medicine, ed 3, New York, Oxford Press. Pasero C, McCaffery M. May be duplicated for use in clinical practice. Although buprenorphine (Buprenex) has some kappa agonist activity, it is known primarily as a partial mu agonist drug. It is referred to as partial because it binds as an agonist at the mu opioid receptors but has limited intrinsic efficacy (Gutstein, Akil, 2006). In the clinical setting, this means that analgesia plateaus as the dose is increased. Buprenorphine also has very high affinity for the mu receptor. It is not readily reversed by opioid antagonists, such as naloxone, which suggests the drug dissociates very slowly from the mu opioid receptor sites (Gutstein, Akil, 2006). The drug is used most often as a maintenance drug for the treatment of addictive disease (Suboxone, Subutex) (see Chapter 13). The type of opioid receptor site and its location determine the effects an opioid drug produces. As mentioned, in addition to producing analgesia, opioid drugs produce a number of other effects, including constipation, nausea and vomiting, sedation, respiratory depression, and urinary retention (Gutstein, Akil, 2006) (see Table 11-1). The main GI effect of opioid drugs is inhibition of GI peristalsis and diminished GI, biliary, and pancreatic secretions, which can lead to constipation and predispose to ileus and other adverse effects as a result of opioid binding to receptors located in the GI tract and CNS (Gutstein, Akil, 2006; Kraft, 2007; Thomas, 2008). Opioid bowel syndrome is described as a “constellation” of undesirable outcomes, including but not limited to constipation (McNicol, Boyce, Schumann, et al., 2008). Nausea and vomiting are the result of opioid binding to receptors located in the fourth ventricle of the brain and direct stimulation of the chemoreceptor trigger zone in the area postrema of the medulla (Freye, 2008). Urinary retention may occur when opioid binding leads to inhibition of the release of acetylcholine (Freye, 2008). Respiratory depression may follow binding in the pontine and ventral medulla of the brainstem (Freye, 2008), and sedation occurs from binding to receptors in the brain (Gutstein, Akil, 2006) (see Chapter 19 for discussion of opioid adverse effects). Absorption is the rate and extent to which a drug leaves its site of administration and moves to plasma or other tissues. A more clinically important concept than absorption is bioavailability, which is the extent to which a dose of a drug reaches its site of action (Buxton, 2006), or how much drug is available for therapeutic effect. Opioid drugs are 100% bioavailable when administered intravenously because they are introduced directly into the systemic circulation. After oral administration, opioids are absorbed from the GI tract and transported by the portal vein to the liver, the primary site of drug metabolism, before they reach systemic circulation. This process reduces the bioavailability of an opioid when administered orally (Buxton, 2006). Oral bioavailability depends on how much of the drug is absorbed in the GI tract and inactivated as it passes through the liver. This is called first pass effect. First pass effect is why the dose of an opioid drug by the oral route must be much larger than by the parenteral route to produce equal analgesia (Buxton, 2006). For example, the bioavailability of morphine when given orally usually is between 20% and 30% because of first pass losses (De Pinto, Dunbar, Edwards, 2006; Gutstein, Akil, 2006; Stevens, Ghazi, 2000) (Figure 11-2). Characteristics of the drug itself also help to determine its bioavailability (Buxton, 2006). A drug’s bioavailability will be decreased if it is a drug for which the liver has a great capacity to metabolize and excrete, such as hydromorphone. When given intravenously, hydromorphone is 100% bioavailable and the recommended starting adult dose for severe pain is 1.5 mg given over a 4-hour period; when given orally, which subjects the drug to a significant first-pass effect, the equianalgesic dose is approximately five times greater at 7.5 mg (see later in this chapter for more on equianalgesia). Metabolites often have properties and characteristics different from their parent drug. Sometimes their pharmacologic actions are indistinguishable from the parent drug, but their biologic activity may be increased, decreased, or eliminated. For example, the major metabolite of morphine, M6G, is analgesic like its parent but is significantly more potent (Buxton, 2006) (see Chapter 13). Cytochrome P450 Enzyme System: Two major hepatic enzyme systems are responsible for metabolism of opioids: CYP450 and, to a lesser extent, the UDP-glucuronosyltransferases (UGTs) (Holmquist, 2009). The UGTs are involved in the formation of glucuronides and the metabolism of hydromorphone, morphine, and oxymorphone. The CYP450 enzyme system is important to the metabolism of codeine, fentanyl, methadone, oxycodone, and oxymorphone (Holmquist, 2009). CYP450 enzymes also are located in other tissues of the body and are the primary enzymes involved in drug metabolism and the production of cholesterol, steroids, prostacylins, and other essential substances (Holmquist, 2009). Some drugs compete with opioids for metabolism by specific enzymes, which may result in drug-drug interactions. Table 11-2 lists potential drug interactions for the CYP2D6 and CYP3A4 enzymes. Box 11-1 provides websites where information can be found regarding drugs that are metabolized by the cytochrome P450 enzyme system and their potential drug-drug interactions. Table 11-2 Potential Drug Interactions for Major Cytochrome P-450 Enzymes CYP3A4 and CYP2D6 From Pasero, C., & McCaffery, M. Pain assessment and pharmacologic management, p. 289, St. Louis, Mosby. Data from Clinical Pharmacology Online. Gold Standard, Inc. Available at http://clinicalpharmacology.com; Cytochrome P450 Interactions ©GlobalRPh Inc. Available at http://www.globalrph.com/cytochrome.htm; Cytochrome P450 and Drug Interactions (Drugs that Induce or Inhibit Various Cytochrome P-450 Systems). Available at http://www.edhayes.com/CYP450-3.html. Pasero C, McCaffery M. May be duplicated for use in clinical practice. Clearance is also a measure of the body’s ability to eliminate a drug from the body. The clearance of a drug depends on the organs of elimination coming in contact with the blood or plasma containing the drug (Buxton, 2006). Because the kidney is the major organ of elimination, renal insufficiency can alter drug clearance. Creatinine clearance analysis is a measure of kidney function and a tool to determine the body’s ability to handle drugs that are primarily eliminated by the kidney. A high creatinine level may be an indication of reduced kidney function and, therefore, a reduced ability to eliminate a drug. Normally, men tend to have higher creatinine levels than women. Aging is associated with decreased body mass and total body water and an increased proportion of body fat which can alter the rates of clearance and elimination (American Geriatrics Society [AGS], 2009; Cook, Rooke, 2003). Box 11-2 provides serum creatinine and creatinine clearance values for all age groups and the formula for calculating creatinine clearance. Box 11-2 Creatinine Values and Formula for Calculating Creatinine Clearance
Physiology and Pharmacology of Opioid Analgesics
Underlying Mechanisms of Opioid Analgesia and Adverse Effects
Endogenous Opioid System
Opioid Receptors
Classes of Opioid Receptor Sites
Opioid Receptors and Adverse Effects
Pharmacologic Concepts
Pharmacokinetics
Absorption, Bioavailability, First Pass Effect, and Solubility
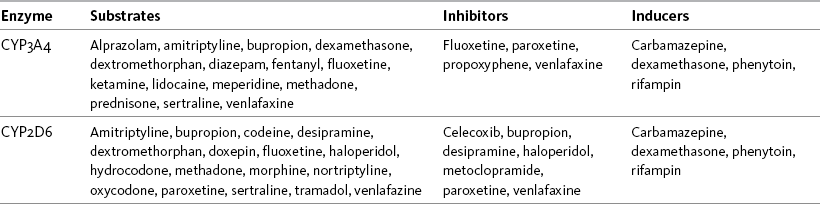
Metabolism, Metabolites, and Prodrugs
Half-Life, Clearance, Steady State, and Accumulation
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree