4.1 Introduction
The pharmacovigilance documents associated with risk evaluation and management are often required components of Marketing Authorization Application (MAA) and New Drug Application (NDA) submissions, and thus could have been included in Chapter 3 (Pharmacovigilance Medical Writing for Marketing Authorization). However, they have been accorded a stand-alone chapter as they are ‘living’ documents, which are continually updated and amended throughout the medicinal product’s post-marketing life, as and when warranted by the emergence of new safety information.
In the EU, the Risk Management Plan (RMP) is a mandated component of the MAA for most medicinal products. The EU RMP outlines all safety information yet to be established for the medicinal product and describes the processes that will be utilized by the company to acquire this information. Furthermore, the RMP outlines the measures that will be applied by the company to minimize the product’s determined risks and how the effectiveness and success of these endeavors will be determined.
In effect, the EU RMP, when submitted at the time of application for marketing authorization, affords both the sponsor and regulatory authority the opportunity to proactively plan all required pharmacovigilance activities before the medicinal product is actually licensed for use by the public. As new safety data is gathered during the medicinal product’s early post-marketing life, this document is updated, either upon the initiative of the Marketing Authorization Holder (MAH) or at the behest of the regulators, to further fine-tune understanding of the product’s safety profile and resulting impact on the benefit-risk balance. As such, the EU RMP prepared at the time of application for marketing authorization remains a live document that will continue to be updated in the course of the medicinal product’s post-marketing life.
In the US, the Risk Evaluation and Mitigation Strategies (REMS) Report can be requested by the Food and Drug Administration (FDA) as part of the registration or authorization process for any medicinal product considered as requiring additional risk management processes. In effect, the REMS Report serves the same purpose in the US as the EU RMP in the EU. Although the REMS Report came into effect as a pharmacovigilance document in 2007, the FDA can also request a REMS Report for medicinal products authorized prior to 2007, if considered necessary due to new safety data. As an alternative, it is also possible for the sponsor or MAH to proactively prepare and submit a REMS Report without a specific request from the FDA.
Preparation of the EU RMP and REMS Report involves a significant degree of pre-submission discussion and correspondence between the applicant (i.e. MAH or sponsor) and the regulators, which serve to guide the applicant on the regulators’ views regarding acceptable risk minimization measures for the medicinal product.
The Benefit-Risk Evaluation Report, which presents a cumulative assessment of the medicinal products’ benefits (in specific indications) weighed against the associated risks, is developed during the pre-authorization period. As with other pharmacovigilance documents utilized in risk evaluation and management, the Benefit-Risk Evaluation Report continues to be updated and amended throughout the medicinal product’s life cycle, and is required for product authorization, with subsequent updating and amendment usually required at other critical decision-making points, such as registration of new indications, license renewal, and, on occasions, of product withdrawal.
A summary of the key pharmacovigilance documents required for risk evaluation and management is presented below (see Figure 1.1):
- EU RMP (EU only);
- REMS Report (US only);
- Benefit-Risk Evaluation reports.
4.2 The EU Risk Management Plan
4.2.1 Regulatory Guidelines and General Principles
Volume 9A, the Rules Governing Medicinal Products in the European Union [1], provides the legal underpinning for the EU RMP, with guidance, including a template published by the ICH (Harmonised Tripartite Guideline Pharmacovigilance Planning E2E) [2] and the European Medicines Agency [3, 4].
The general principles of the EU RMP are summarized in Table 4.1.
Table 4.1 General principles of the EU Risk Management Plan.
| Principle | Description |
| Scope of Data | The EU RMP encompasses numerous data, including clinical, non-clinical, epidemiological, and literature data. |
| Risk Assessment | The initial component of the EU RMP; comprises the Safety Specification (describing important identified risks, potential risks, and missing information) and Pharmacovigilance Plan (describing standard pharmacovigilance processes and measures to further examine safety concerns described in the Safety Specification). |
| Risk Minimization | The second component of the EU RMP; comprises assessment of the need for further pharmacovigilance activities and methods for monitoring the effectiveness of the implemented measures. |
| Related Safety Documents | The safety data presented in the EU RMP should be consistent with that presented in the other related safety documents (e.g. PSUR, DSUR, Investigator’s Brochure, and product labeling). |
| DSUR = Development Safety Update Report; EU = European Union; PSUR = Periodic Safety Update Report; RMP = Risk Management Plan | |
4.2.2 When are EU Risk Management Plans Prepared?
EU RMPs can be prepared and submitted at any time during the medicinal product’s pre- and post-marketing life cycle. EU RMPs are prepared or updated on the following occasions:
- at the time of the initial MAA (i.e. at the time of product registration/authorization);
- application for registration of new indications or changes to approved use (e.g. new dose, new route of administration, or a modified manufacturing process for biotechnology products);
- authorization of treatment for a special treatment population (e.g. paediatrics and the elderly);
- identification of a new safety concern;
- upon request from the relevant competent authority;
- within 60 days of important pharmacovigilance or risk minimization activity milestones being achieved or availability of new data from studies.
4.2.3 Data Sources for the EU Risk Management Plan
A summary of the data required for preparation of EU RMPs and the company departments usually responsible for the provision of these data are presented in Table 4.2. As noted with other documents, the precise designations of the departments/functions may vary from company to company.
Table 4.2 Source data for the EU Risk Management Plan.
| EU RMP Data | Data Source |
| Non-clinical Data | Non-clinical R&D; data relating to all pertinent safety issues not yet resolved in the clinical setting (e.g. toxicity, drug interactions, and pharmacology related complications). |
| Clinical Data – Clinical trials – Marketplace | Clinical Operations; patient exposure data in studies Drug Safety (Pharmacovigilance) or Sales and Marketing; post-marketing exposure data. |
| Regulatory Actions Taken | Regulatory Affairs |
| Adverse Events/Adverse Experience | Drug Safety (Pharmacovigilance)In addition: – Non-clinical R&D; identified and potential drug interactions. – Clinical and Non-clinical R&D; epidemiology of indications and important adverse events. |
| Pharmacovigilance Plan | Drug Safety (Pharmacovigilance) |
| Literature | Medical Information/Scientific Information Services |
| EU = European Union; R&D = Research and Development | |
4.2.4 Review of the EU Risk Management Plan
As with all other pharmacovigilance documents, the preparation of the EU RMP involves contributions from a multidisciplinary team, which is tasked with participation in document planning, provision of data from their respective departments, and review and approval of the RMP. A summary of the team that should be involved in preparation of the EU RMP is presented in Table 4.3.
Table 4.3 The EU Risk Management Plan review team.
| Reviewer | Key Areas of Responsibility/Sections to Review |
| Drug Safety Physician | The whole EU RMP |
| EU Qualified Person/Director of Safety | The whole EU RMP |
| Medical Affairs | The whole EU RMP |
| Clinical and Non-clinical R&D | Clinical: the whole EU RMP Non-clinical: Section 1: Safety Specification |
| Regulatory Affairs | Section 1.4.2: Regulatory Actions Taken |
| Quality Control | The whole EU RMP |
| EU = European Union; R&D = Research and Development; RMP = Risk Management Plan | |
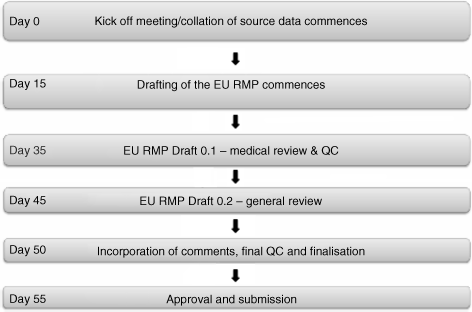
4.2.5 A Timeline – Planning for the EU Risk Management Plan
As a general rule, preparation of an EU RMP can be divided into the following four key activities:
- RMP planning and collation of source data;
- writing of the draft EU RMP;
- review of the EU RMP;
- QC, finalization, and approval of the EU RMP.
An example timeline appropriate for preparation of the EU RMP is presented in Figure 4.1.
4.2.6 Generic Model of the EU Risk Management Plan
A generic model EU RMP template, based on guidelines and format recommended by the European Medicines Agency [3] is presented in this section.
| Abbreviations |
| Product Information |
| 1 Safety Specification |
| 1.1 Non-clinical |
| 1.2 Clinical |
| 1.2.1 Limitations of the Human Safety Database |
| 1.3 Populations not Studied in the Pre-authorization Phase |
| 1.4 Post-authorization Experience |
| 1.4.1 Post-authorization Usage Data |
| 1.4.2 Regulatory Actions Taken |
| 1.5 Adverse Events/Adverse Reactions |
| 1.5.1 Newly Identified Safety Concerns since the Last Submitted EU RMP |
| 1.5.2 Important Identified and Potential Risks (including Newly Identified Risks) |
| 1.6 Identified and Potential Interactions with other Medicinal Products, Food, and other Substances |
| 1.7 Epidemiology of the Indications and Important Adverse Events |
| 1.8 Pharmacological Class Effects |
| 1.9 Additional EU Requirements |
| 1.9.1 Potential for Overdose |
| 1.9.2 Potential for Transmission of Infectious Agents |
| 1.9.3 Potential for Misuse for Illegal Purposes |
| 1.9.4 Potential for Off-Label Use |
| 1.9.5 Potential for Off-Label Paediatric Use |
| 1.10 Summary – Ongoing Safety Concerns |
| 2 Pharmacovigilance Plan |
| 2.1 Routine Pharmacovigilance Practices |
| 2.2 Summary of Safety Concern and Planned Pharmacovigilance Actions |
| 2.3 Detailed Action Plan for Specific Safety Concerns |
| 2.4 Overview of Study Protocols for the Pharmacovigilance Plan |
| 2.5 For Updates to the EU RMP |
| 2.6 Summary of Outstanding Actions, including Milestone |
| 3 Evaluation of the Need for Risk Minimization Activities |
| 3.1 Summary Table of Planned Actions |
| 3.2 Potential for Medication Errors |
| 4 Risk Minimization Plan |
| 5 Summary of EU Risk Management Plan |
| 6 Contact Person Details |
| Annexes |
| Abbreviation | Definition |
| ATC | Anatomical Therapeutic Chemical |
| CCSI | Company Core Safety Information |
| EEA | European Economic Area |
| EU | European Union |
| EUQPPV | EU Qualified Person for Pharmacoviglance Professional |
| INN | International non-proprietary name |
| MAA | Marketing Authorization Application |
| MAH | Marketing Authorization Holder |
| MedDRA | Medical Dictionary for Regulatory Activities |
| PD | Pharmacodynamic |
| PK | Pharmacokinetic |
| PSUR | Periodic Safety Update Report |
| PT | Preferred term |
| RMP | Risk Management Plan |
| SmPC | Summary of Product Characteristics |
| Note: The table needs to be expanded and completed as required. | |
| Invented name of the medicinal product (product short name) | Add details of product brand name. |
| Active substance(s) (INN or common name): | Add details of the product generic name. |
| Pharmaco-therapeutic group (ATC Code): | Add products ATC code. |
| Medicinal product code (From Eudra Vigilance) | Add details. |
| Authorization procedure(s) (central, mutual recognition, decentralized, national) | Add details of the product’s registration procedure in the EU. |
| Name of Marketing Authorization Holder or Applicant | Add company details. |
| Date and country of first authorization worldwide | Complete as appropriate. |
| Date and country of first launch worldwide | Complete as appropriate. |
| Date and country of first authorization in the EEA | Complete as appropriate – if different from above. |
| Date and country of first launch in the EEA | Complete as appropriate – if different from above. |
| Data lock point for EU RMP | Add date of data cut-off. |
| Version | Add version number (i.e. version 1.0 for the first final version, and versions 2.0, 3.0, etc. on subsequent updates). |
| Brief description of product (chemical class, mode of action, etc.) | This section should include a brief description of the product, including the active ingredient, therapeutic class, and mechanism of action/pharmacology. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Indication(s) | Add details of the proposed indications if this is the first RMP prepared as part of the MAA dossier, or, details of registered indications if this is an update of an existing RMP for marketed products. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Dosage | This section should present details of the proposed (or registered) doses for the medicinal product. If the product is proposed (or approved) for more than one indication, this section should be separated into subsections according to indication and dosage details presented for each indication. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Pharmaceutical form(s) and strength(s) | The physical form in which the medicinal product is presented for use, and the amount of active substance in each unit is described in this section. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ATC = Anatomical Therapeutic Chemical; EEA = European Economic Area; EU = European Union; INN = International non-proprietary name; MAA = Marketing Authorization Application; RMP = Risk Management Plan | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
- identified risks;
- potential risks;
- unknown or missing information.
- toxicity (e.g. repeat-dose toxicity, reproductive/development toxicity, nephrotoxicity, hepatotoxicity, genotoxicity, and carcinogenicity);
- general pharmacology (e.g. cardiovascular, nervous system);
- drug interactions.
| Safety Concern from Non-clinical Studies | Relevance to Human Usage |
| Repeat-Dose Toxicity | Complete as appropriate. |
| Reproductive Toxicity | Complete as appropriate. |
- clinical trial exposure;
- epidemiological study exposure;
- post-marketing exposure.
- type of study (e.g. blinded randomized studies, open studies, observational studies, overall exposure);
- age;
- gender;
- doses administered;
- duration of treatment.
- children;
- elderly patients (usually defined as aged ≥65 years);
- pregnant or breast-feeding women;
- patients with organ dysfunction (e.g. hepatic or renal impairment);
- patient with disease severity different from the clinical study population;
- patient populations with relevant genetic polymorphism;
- patient populations from varying ethnic/racial origins.
| Safety Concern #1 | |
| Details: | Add summary of safety concern. |
| Source: | Add summary of data used to identify the safety issue. |
| Implications for product literature | Add a summary of modifications proposed for product literature (e.g. SmPC, CCSI, and PI) as a result of the newly identified safety issue. |
| New studies proposed in the Pharmacovigilance Plan | Yes/No |
| New risk minimization actions proposed | Yes/No |
| Safety Concern #2 | |
| Expand table as required | |
| CCSI = Company Core Safety Information; PI = Package Insert; SmPC = Summary of Product Characteristics | |
| Identified Risk of <x> | Add MedDRA PT term |
| Seriousness/outcome | Complete as appropriate (e.g., the proportion of patients with outcomes recorded as: fatal, recovered with or without sequelae, not recovered, hospitalized). |
| Severity and nature of risk | Include a tabulated summary of severities (e.g., % of mild, moderate or severe). |
| Frequency with 95% CI | Present the relative incidence as well as incidence compared to placebo/comparator treatments. |
| Background incidence/prevalence | Present the background incidence and the general prevalence in the target population(s). |
| Risk groups or risk factors | Describe use, including doses and patient susceptibility or other risk factors. |
| Potential mechanisms | Describe mechanisms. |
| Preventability | Present available data on the potential to predict and prevent the risk(s). |
| Potential public health impact of safety concern | If feasible include the number of patients expected to be affected, hospitalizations, fatal outcomes etc. |
| Evidence source | Present data sources by cross-referencing to CTD modules or specific studies and other reports (e.g., PSURs). |
| Regulatory action taken | Specify the country and type of action taken. |
| CTD = Common Technical Documentation; MedDRA = Medical Dictionary for Regulatory Activities; PSUR = Periodic Safety Update Report; PT = preferred term, | |
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


