3.1 Introduction
The process of pre-authorization clinical development is considered complete when the sponsor has collated sufficient evidence of efficacy and safety for the investigated medicinal product. Thereafter, it is time to compile all the accrued non-clinical and clinical data into dossiers for submission to the regulatory authorities responsible for granting marketing authorization.
In the EU and US regions, dossiers prepared in pursuit of marketing authorization (referred to as Marketing Authorization Application [MAA] and New Drug Application [NDA] in the EU and US, respectively) use a standardized format known as the Common Technical Documentation (CTD). This comprises a set of documents devoted to the analysis of data relating to the quality, safety, and efficacy of the medicinal product.
Documents dedicated to safety analyses represent a significant component of the CTD dossier. Routinely, qualified personnel at the regulatory authority meticulously review all the safety data collated in the course of the clinical development program, to ensure that the medicinal product has an acceptable safety profile and is well tolerated in the proposed indication, treatment dose(s), route(s) of administration, and treatment population.
Whereas the Summary of Clinical Safety (SCS), also referred to as CTD Module 2.7.4, is the main analysis of safety for the EU region, submissions to the US require the SCS and two other documents not mandated in the EU, namely, the Integrated Summary of Safety (ISS) and the 120-Day Safety Update Report.
Although the SCS and ISS both present analyses of safety data gathered during the clinical development program, they differ in that the SCS is a clinical summary, in which summaries of safety data can be presented by pooling studies (if study designs are sufficiently similar to permit pooling) or by individual study. As such, the SCS can be prepared without pooling studies but by simply relying on data as presented in the individual clinical study reports. In contrast, the ISS is not a clinical summary but an integrated overall analysis, which presents safety data that has undergone statistical analysis after data from the individual studies have been pooled into a single database. The rationale for this is that analysis from a pooled database increases the likelihood of detecting potential treatment-related adverse effects that may occur at low frequencies. Thus, the ISS summarizes the same data that are presented in the SCS, but affords a level of scrutiny greater than that achieved through analysis of individual study data. Evaluation of data from a single integrated database also permits better comparison of treatment-emergent adverse event (TEAE) rates between placebo-treated subjects and actively-treated subjects to determine the TEAEs that represent adverse drug reactions (ADRs) (i.e. related TEAEs). This therefore reduces reliance on the investigator’s opinion of causality.
Safety data presented in the SCS and/or ISS are of critical importance, as this forms the basis for the medicinal product’s labeling in the Summary of Product Characteristics (SmPC) (for the EU) and United States Package Insert (USPI) (for the US), representing the information provided to prescribers and other healthcare professionals in respect of the safe and recommended use of the product.
As the name implies, the 120-day Safety Update Report is mandated for submission to the Food and Drug Administration (FDA) 120 days after submission of the NDA, and is intended to provide a summary update of any new safety data gathered by the sponsor since the data cut-off for the NDA submission documents, which could have been as far back as 6 months prior to the NDA submission date. In effect, the 120-day Safety Update Report could represent almost 1 year’s worth of new safety data, which needs to be reviewed by the authorities to ensure there has been no change in the product’s recorded safety profile. This is particularly important for medications intended for long-term treatment.
In summary, the following pharmacovigilance documents are required for marketing authorisation (see Figure 3.1):
- SCS (EU and US);
- ISS (US only);
- 120-day Safety Update Report (US only).
3.2 The Summary of Clinical Safety
3.2.1 Regulatory Guidelines and General Principles
The purpose of the SCS is to present safety data clearly describing the safety profile of the medicinal product for which the sponsor seeks marketing approval. Guidance on the required content and format of the SCS is provided in the Notice to Applicants – Medicinal Products for Human Use [1]. The general principles underlying presentation of safety data in the SCS are summarized in Table 3.1.

Table 3.1 General principles for the SCS.
| Principle | Description |
| Scope of Data | The SCS comprises safety data from the following: – All studies for the medicinal product undertaken in the clinical development program – Global post-marketing data (if the investigated medicinal product is also marketed) – Published literature |
| Safety Population | As with ICH E3 clinical study reports, the safety population includes all subjects exposed to at least one dose of the investigated medicinal product in the clinical studies. Exclusion of any subject exposed to study treatment from the safety population must be justified. |
| TEAEs | TEAEs are defined as AEs that were not present at study baseline and occurred after administration of the medicinal product, or AEs that were present at study baseline but increased in intensity/severity after administration of the medicinal product. |
| AE = adverse event; ICH = International Conference on Harmonisation; SCS = Summary of Clinical Safety; TEAE = treatment-emergent adverse event | |
3.2.2 Data Sources for the SCS
Source data required by the PV Medical Writer before preparation of the SCS can commence is presented in Table 3.2, with details of the departments/functions usually tasked with responsibility for provision of such data to the PV Medical Writer.
Table 3.2 Source data for the SCS.
| SCS Data | Data Source |
| Safety Data from the Clinical Development Program | Medical Writing – All clinical study reports for studies undertaken in the clinical program – Tables, figures, and listings from any pooled studies (if applicable) – Study reports relating to PK and PD studies – Study reports relating to reproductive toxicology, etc. |
| Post-Marketing Safety Data | Drug Safety (Pharmacovigilance) – ICH E2C line listings – Summary tabulations – CIOMS reports for all reported cases |
| Post-Marketing Sales Data | Sales and Marketing |
| Literature | Medical Information/Scientific Information Services |
| CIOMS = Council for International Organizations of Medical Sciences; ICH = International Conference on Harmonisation; PD = pharmacodynamic; PK = pharmacokinetic; R&D = Research and Development; SCS = Summary of Clinical Safety | |
3.2.3 Review of the SCS
As with all safety documents, preparation of the SCS is a team effort requiring contribution (i.e. in terms of data provision as well as document review and approval) from a number of different departments. It is important to remember that the specific designation of these departments will vary from company to company. Again, the PV Medical Writer’s role in the project is central, as responsibility for coordinating the entire process and ensuring that all participants fulfil their roles rests squarely on his/her shoulders.
A typical team involved in the preparation of the SCS is presented in Table 3.3.
Table 3.3 The SCS review team.
| Reviewer | Key Areas of Responsibility |
| Statistics and Data Management | Ensures correct interpretation of statistical data (either pooled or non-pooled) are used. |
| Regulatory Affairs | Ensure that conclusions reached in the SCS with respect to the medicinal product’s safety profile are consistent with the product’s labeling, whether draft labeling in preparation (for pre-authorized products) or existing labeling (for marketed products) that may also be undergoing simultaneous updating. |
| Drug Safety Physician | Ensure that data is presented objectively and appropriate conclusions are drawn from the presented safety data. |
| Qualified Person (EU)/Director of Drug Safety | Ensure that data is presented objectively and appropriate conclusions are drawn from the presented safety data. |
| Medical Affairs | Ensure that data is presented objectively and appropriate conclusions are drawn from the presented safety data. |
| Regulatory Advertising, Labeling and Promotions | Ensure that conclusions reached in the SCS with respect to the medicinal product’s safety profile are consistent with the product’s labeling, whether draft labeling in preparation (for pre-authorized products) or existing labeling (for marketed products) that may also be undergoing simultaneous updating. |
| QC | Ensure consistency between cited data and source documents, and also checks for grammar, punctuation, and use of language. |
| EU = European Union; SCS = Summary of Clinical Safety; QC = quality control | |
It is customary to have one team for the initial stage of document review, followed by the team review meeting, during which all comments or issues that require discussion for a consensus position to be reached can be dealt with. The subsequent draft of the document is reviewed by the heads of each department/function, which essentially serves as the final review of the document before it is forwarded to the senior management team for document approval prior to submission to the authorities.
3.2.4 A Timeline – Planning for the SCS
Preparation of a SCS involves a number of key activities:
- planning and collation of source data;
- writing and reviewing the draft SCS;
- quality control (QC) activities;
- approval and submission.
In general, preparation of the SCS, from the planning stage to finalization, through approval and submission can take up to 4 months, given the time to be allowed for multidisciplinary team reviews and review meetings before each draft of the document can proceed to the next stage (Figure 3.1).
3.2.4.1 Planning for the SCS and Collation of Source Data
Preparation activities for the SCS are undertaken during weeks 1–4, including collation of all study reports (clinical and non-clinical) from which data for the SCS is to be generated. In addition, tables and listings from any pooled analyses are prepared by the statistical and data management functions, reviewed by the PV Medical Writer, and finalized. A literature search pertinent to the medicinal product and therapeutic area is also undertaken, and safety and patient exposure data collected from the post-marketing experience (if the product is also marketed).
3.2.4.2 Writing and Reviewing of the Draft SCS
Preparation of Draft 0.1 can take approximately 3 weeks. The completed draft SCS is circulated to the review team (as outlined in Table 3.3) for their review and comment, the duration of which can vary from 1–2 weeks, depending on company policy and the team members’ competing priorities. Although some companies still review documents using track changes in Microsoft Word/Office, a popular method of team review is the Documentum-based electronic document management system, which companies are increasingly using for document review, retention of review comments, and proof of review, as well as being the ultimate repository of the finalized documents.
It is usual to schedule a team review meeting after the review period, as an opportunity for outstanding or contradictory comments from different team members to be discussed by the team.
Preparation of Draft 0.2 commences after the team review meeting, and the process is repeated by way of a second team review and team review meeting. Following this second review cycle, Draft 0.3 is submitted for QC against all source documents used, the company’s style guide and document template. After incorporation of QC comments, Draft 0.4 should now be in a final form that can be submitted for approval.
It is to be noted that this example timeline can be modified according to the team’s needs and availability, and should indeed form part of the discussions during the SCS planning stage. In addition, the number of SCS drafts can be increased if considered appropriate.
3.2.5 Generic Model of the SCS
In this generic model of the SCS, each section is examined and a summary of the data to be presented in the section is presented below.
| List of Abbreviations |
| List of Tables |
| List of Figures |
| 1 Exposure to Drug |
| 1.1 Overall Safety Evaluation Plan and Narratives of Safety Studies |
| 1.2 Overall Extent of Exposure |
| 1.3 Demographic and Other Characteristics of Study Population |
| 2 Adverse Events |
| 2.1 Analysis of Adverse Events |
| 2.1.1 Common Adverse Events |
| 2.1.2 Analysis of TEAEs by Dose |
| 2.1.3 Analysis of TEAEs by Severity |
| 2.1.4 Analysis of TEAEs by Relatedness |
| 2.1.5 Deaths |
| 2.1.6 Other Serious Adverse Events |
| 2.1.7 Other Significant Adverse Events |
| 2.1.8 Analysis of Adverse Events by Organ System and Syndrome |
| 2.2 Narratives |
| 3 Clinical Laboratory Evaluations |
| 4 Vital Signs, Physical Findings, and Other Observations Related to Safety |
| 5 Safety in Special Patient Groups and Situations |
| 5.1 Intrinsic Factors |
| 5.2 Extrinsic Factors |
| 5.3 Drug Interactions |
| 5.4 Use in Pregnancy and Lactation |
| 5.5 Overdose |
| 5.6 Drug Abuse |
| 5.7 Withdrawal and Rebound |
| 5.8 Effects on Ability to Drive or Operate Machinery or Impairment of Mental Ability |
| 6 Post-marketing Data |
| 7 References |
| 8 Appendices |
| Abbreviation | Definition |
| ADR | Adverse drug reaction |
| AE | Adverse event |
| CCSI | Company Core Safety Information |
| CTD | Common Technical Documentation |
| EU | European Union |
| MedDRA | Medical Dictionary for Regulatory Activities |
| PSUR | Periodic Safety Update Report |
| PD | Pharmacodynamic |
| PK | Pharmacokinetic |
| PT | Preferred term |
| SCE | Summary of Clinical Efficacy |
| SCS | Summary of Clinical Safety |
| SmPC | Summary of Product Characteristics |
| TEAE | Treatment-emergent adverse event |
| USPI | United States Package Insert |
| Note: The table needs to be expanded and completed as required. | |
- healthy volunteers;
- subjects from the targeted patient population;
- safety data from the market place (if the product is already marketed for other uses).
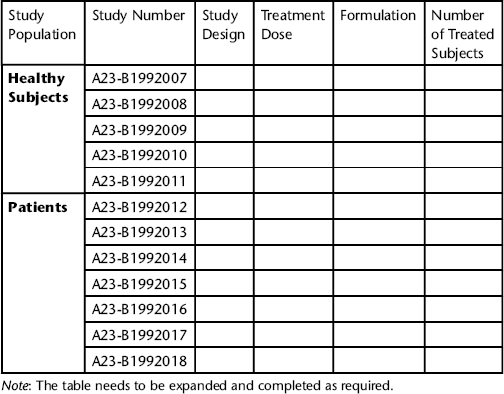
- the number of studies (with description of study design) undertaken and description of study population;
- the total number of subjects exposed to treatment (including dose[s] and route[s] of administration);
- description of study categorization (i.e. pivotal studies, supporting studies).
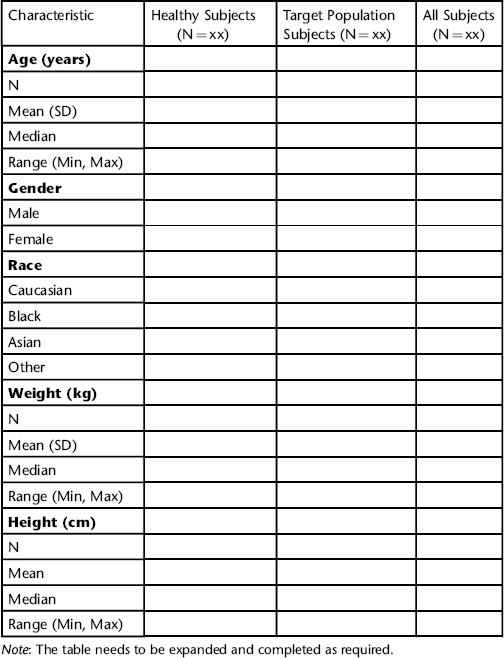
- overall exposure (i.e. for the total subject population);
- healthy volunteers/healthy subjects;
- subjects representative of the target patient population;
- post-marketing exposure.
- the total subject population;
- healthy volunteers/healthy subjects;
- subjects representative of the target patient population;
- different indications.
- use of concomitant medications;
- concomitant illnesses.
- geographical location;
- disease severity;
- impaired renal function;
- impaired hepatic function.
| Placebo (N = xx) | Product X (N = xx) | |
| Number (%) of subjects with: | ||
| At least one AE | ||
| At least one TEAE | ||
| At least one serious TEAE | ||
| At least one related serious TEAE | ||
| At least one TEAE leading to treatment withdrawal | ||
| At least one severe TEAE | ||
| At least one related TEAE | ||
| AE = adverse event; TEAE = treatment-emergent adverse event | ||
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


