5.1 Introduction
Once a medicinal product has been approved for marketing, the marketing authorization holder (MAH) has an obligation to periodically submit a number of pharmacovigilance reports to relevant regulatory authorities in the regions where the product is marketed. These periodic reports are a concise analysis of new safety data gathered since marketing approval, and allow for the continued surveillance of the product’s safety, as well as continued appraisal of potential new risks and changes to the product’s benefit-risk profile, all of which act to ensure the public’s safety.
In this context, it is important to appreciate that safety is not an absolute concept, and medicinal products that have successfully passed through the long drug development process still need to be monitored once they have gained entry to the marketplace. This is due to the nature of the clinical trials conducted during the drug development process, which, for reasons of practicality, generally involve a limited number of carefully selected (and carefully monitored) participants and run for limited durations of time. The consequence of these practical constraints is that, although certain types of side effects are detected during clinical development, others only ever come to light once the product is in the marketplace, being used by a much larger patient population outside the confines of the tightly controlled and regulated clinical trial environment. Viewed in this light, the need and importance of continued surveillance facilitated by the effort that informs the preparation of the periodic and mandated post-marketing pharmacovigilance documents is inarguable.
As such, these post-marketing pharmacovigilance documents allow both the MAH and the regulatory authority periodic and continued reviews of the product’s safety profile by presenting:
- a summary of any changes in the product’s worldwide registrations and any actions taken for reasons of safety;
- a summary of all safety data collated in the review period, and a cumulative review of data collected since marketing authorization;
- a conclusion indicating whether the product’s labeling and promotional literature require updating, or sufficiently describe the known risks associated with product use.
The Periodic Safety Update Report (PSUR) is the main post-marketing pharmacovigilance document required throughout the product’s post-marketing life in the EU.
In the US, submission of a New Drug Application (NDA) Periodic Adverse Drug Experience Report (PADER) fulfils the same purpose as a PSUR in the EU. However, it is worth noting that, for reasons of practicality, if an EU PSUR already exists for a given product, it is common practice for the Food and Drug Administration (FDA) to permit submission of the EU PSUR (through application for a waiver) in place of a US PADER, thereby relieving the MAH of the burden of preparing two separate periodic reports for the same product.
A summary of the general structure of a PSUR and PADER is presented in Table 5.1, to highlight the key differences in the format of these documents.
Table 5.1 The EU PSUR and US PADER – key differences in content.
| EU PSUR | US PADER |
| Worldwide Registration Status | Summary analysis of submitted 15-day reports |
| Summary of regulatory or MAH actions taken for reasons of safety | Summary tabulations of individual events by SOC (from the 15-day reports) |
| Changes to RSI | MedWatch forms for cases not submitted as 15-day reports |
| Individual case histories (from all cases received by the MAH during the review period) | Actions taken for reasons of safety |
| Studies Overall evaluation of safety (including serious unlisted reactions; non-serious unlisted reactions; increased reporting of listed reactions) | Note: US PADERs exclude AEs that occurred outside the US and literature case reports (except the 15-day reports). |
| AE = Adverse Event; EU = European Union; MAH = Marketing Authorization Holder; PADER = Periodic Adverse Drug Experience Report; PSUR = Periodic Safety Update Report; RSI = Reference Safety Information; SOC = System Organ Class; US = United States | |
In addition to timely submission of PSURs/PADER according to the applicable scheduling and periodicity, additional submission of PSURs with PSUR Addendums and Summary Bridging Reports (SBRs) is required in cases of:
- license renewals;
- registrations of the product in new countries;
- registration of new indications in markets where the product is already registered.
In such cases, a PSUR Addendum report is required to accompany submission of a 6-monthly or 1-year PSUR that is more than 3 months outside Data Lock Point (DLP), or a 3-year PSUR that is more than 6 months outside the DLP. A SBR is required for submissions that contain more than one PSUR, or contain one PSUR and a PSUR Addendum. Further to the PSURs, PSUR Addendums, and SBRs, the MAH may need to prepare ad-hoc safety reviews and update Risk Management Plans (RMP) and Benefit-Risk Evaluation Reports throughout the product’s post-marketing life. RMPs and Benefit-Risk Evaluation Reports are discussed in Chapter 4 (Pharmacovigilance Medical Writing in Risk Evaluation and Management) and ad-hoc safety summaries in Chapter 6 (The Ad-hoc Safety Review and Response to Questions Document).
To summarize, the following pharmacovigilance documents are required in the post-marketing life of a medicinal product (see Figure 1.1.):
- PSUR/PADER;
- PSUR Addendum;
- SBR;
- RMP and Benefit-Risk Evaluation Reports;
- Ad-hoc safety reviews.
5.2 The EU Periodic Safety Update Report
5.2.1 Regulatory Guidelines and General Principles
The content of EU PSURs, and the legally binding obligations undertaken by the MAH with respect to the submission of post-marketing pharmacovigilance documents, are primarily governed by Volume 9A, the Rules Governing Medicinal Products in the European Union [1], published by the European Commission after consultation with the European Medicines Agency (EMA), member states, and interested parties, in fulfilment of Article 106 of Directive 2001/83/EC and Article 26 of Regulation (EC) No. 726/2004. The requirement for PSUR submission is an integral condition of the granted marketing authorization, and failure to comply can result in license withdrawal or suspension.
In addition to Volume 9A, the Council for International Organizations of Medical Sciences (CIOMS), which was established through an alliance between the World Health Organization and United Nations Educational, Scientific and Cultural Organization (as an arena for scientific experts, pharmaceutical companies, and government bodies to develop guidelines regarding the exchange of safety data between pharmaceutical companies and the government entities charged with industry regulation), established guidelines regarding the content, format, and submission of PSURs. These recommendations from the CIOMS II Working Group [2] were adopted, and indeed largely used as the template for the International Conference on Harmonisation (ICH) guideline for the preparation of PSURs (ICH E2C [R1] [3] which harmonizes the requirements for periodic post-marketing reporting in PSURs across all three ICH regions [i.e. EU, US, and Japan]).
The key general principles for EU PSURs, as defined by Volume 9A and ICH E2C, are summarized in Table 5.2.
Table 5.2 General principles for the EU PSUR.
| Principle | Description |
| Scope of Data | The EU PSUR should encompass the following: – A concise analysis of adverse events reported during the review period – A cumulative analysis of adverse events reported since the IBD – Other relevant information (e.g. follow-up information, RMPs, and Benefit-Risk Evaluation reports) – Reports describing lack of efficacy (especially when relating to safety concerns) – Increased frequency of known adverse events – Medical opinion on the product’s prevailing safety profile |
| One Active One PSUR | All data for all indications, dosage forms, and routes of administration registered for a medicinal product with the same active ingredient that is registered to the same MAH (even if marketed through a licensing partner) should be presented in a single PSUR. |
| RSI | The RSI is the official summary of the product’s known safety profile, against which all reported events can be compared, to determine whether the new safety data is consistent with the adverse effects normally expected for the product in question. |
| IBD | The date the first marketing authorization of the medicinal product was granted to the MAH. |
| DLP | The date of data cut-off for any PSUR review period. |
| DLP = Data Lock Point; EU = European Union; IBD = International Birth Date; MAH = Marketing Authorization Holder; PSUR = Periodic Safety Update Report; RMP = Risk Management Plan; RSI = Reference Safety Information | |
5.2.2 Scheduling and Periodicity – When are EU PSURs Prepared?
EU regulations require PSURs to be submitted for approved drugs in accordance with the scheduling summarized in Figure 5.1.

In addition to the above International Birth Date (IBD)-based scheduling of EU PSUR submissions, the MAH can also be required to submit a PSUR outside this schedule upon a request by a regulatory authority.
5.2.2.1 Exceptions to Standard EU PSUR Scheduling
There are a number of situations when the clock on PSUR scheduling described above can be re-started, and a product that had progressed to yearly or 3-yearly submission intervals can be taken back to the initial 6-monthly schedule of the first 2 years. These include:
- registration of a new indication or treatment population;
- registration of a new dose or method of administration;
- an updated RMP requiring specific monitoring of a safety concern;
- a new active substance that is a different salt/ester of the original active ingredient;
- a new excipient lacking an established safety profile.
5.2.3 Data Sources for the EU PSUR
A summary of the data required before embarking on PSUR preparation is presented in Table 5.3. The departments responsible for supplying each piece of source data will invariably be known by varying titles in different companies, but will largely follow the same pattern. It is also worth noting that different competent authorities within the EU may ask for additional line listings and summary tabulation outside the standard ICH E2C (R1) output presented in Table 5.3.
Table 5.3 Source data for the EU PSUR.
| PSUR Data | Data Source |
| Safety Data – Line listings – Summary Tabulations | Drug Safety (Pharmacovigilance); the following line listings and summary tabulations are required for the EU region: – ICH E2C line listing of medically confirmed serious and non-serious unlisted cases – ICH E2C line listing of medically confirmed non-serious listed cases – ICH E2C line listing of consumer cases – Summary tabulation of medically confirmed serious and non-serious unlisted cases – Summary tabulation of medically confirmed non-serious listed cases – Summary tabulation of consumer cases – Cumulative summary tabulation of all medically confirmed serious unlisted cases from the IBD to the PSUR DLP – ICH E2C line listing of follow-up reports – Late-breaking information – CIOMS reports for all reported cases |
| Patient Exposure – Marketplace – Clinical Trials | Sales and Marketing Clinical Operations |
| RSI | Drug Safety (Pharmacovigilance) & Regulatory Affairs |
| Worldwide Marketing Authorization Status | Regulatory Affairs |
| Regulatory Authority and MAH Actions Taken for Reasons of Safety | Regulatory Affairs & Drug Safety (Pharmacovigilance) |
| RMP & Benefit-Risk Evaluation Report Updates | Regulatory Affairs & Drug Safety (Pharmacovigilance) |
| Literature | Medical Information/Scientific Information Services |
| PSUR Assessment Report | Regulatory Affairs |
| Clinical Studies Non-clinical Studies – Toxicology – Pharmacology | Clinical Operations and Medical Writing Non-clinical R&D |
| CIOMS = Council for International Organizations of Medical Sciences; DLP = Data Lock Point; EU = European Union; IBD = International Birth Date; ICH = International Conference on Harmonisation; MAH = Marketing Authorization Holder; PSUR = Periodic Safety Update Report; R&D = Research & Development; RMP = Risk Management Plan; RSI = Reference Safety Information | |
5.2.4 Review of EU PSURs
The preparation of an EU PSUR is a multidisciplinary team effort and personnel from several departments will be tasked with provision of source data, review, and approval of PSUR sections pertinent to their respective areas of responsibility.
The team involved in a representative PSUR review and approval is presented in Table 5.4.
Table 5.4 The EU PSUR review team.
| Reviewer | Key Areas of Responsibility/Sections to Review |
| Regulatory Affairs | Section 1: Introduction Section 2: Worldwide Marketing Authorization Status Section 3: Update of Regulatory Authority or MAH Actions Taken for Reasons of Safety Section 4: Changes to Reference Safety Information Section 8: Risk Management Programs and Benefit-Risk Evaluation Reports |
| Drug Safety Physician | The whole PSUR, particularly medical conclusions drawn from included cases of adverse events in the following sections: Section 6: Presentation of Individual Case Histories Section 8: Other Information (incl. Efficacy-Related Information, Risk Management Program, and Benefit-Risk Evaluation reports) Section 9: Overall Safety Evaluation |
| Medical Affairs | Medical conclusions drawn from included cases of adverse events in the following sections: Section 6: Presentation of Individual Case Histories Section 9: Overall Safety Evaluation |
| Qualified Person | The whole PSUR, particularly medical conclusions drawn from included cases of adverse events in the following sections: Section 6: Presentation of Individual Case Histories Section 8: Other Information (incl. Efficacy-Related Information, Risk Management Program, and Benefit-Risk Evaluation reports) Section 9: Overall Safety Evaluation |
| Quality Control | The whole PSUR |
| EU = European Union; MAH = Marketing Authorization Holder; PSUR = Periodic Safety Update Report | |
5.2.5 A Timeline – Planning for the EU PSUR
Preparation of the PSUR comprises four key activities, namely:
- PSUR planning and collation of source data;
- writing of the draft PSUR;
- review of the draft PSUR;
- QC activities and PSUR finalization.
EU PSURs have to be submitted to the appropriate regulatory authority by Day 60 after the data cut-off date, referred to as the DLP. Therefore, these planning, writing, and review activities, involving an interdisciplinary team, need to be completed within a 60-day timeline. As a result, the PSUR writing process needs to be initiated some time before the DLP, as outlined in the example timeline presented in Figure 5.2.

5.2.5.1 PSUR Planning and Collation of Source Data
At Day -30, draft ICH E2C line listings for inclusion in the PSUR (as listed in Table 5.3) need to be prepared and reviewed. Draft line listings are initially reviewed by the responsible Pharmacovigilance Officer, to ensure that all included adverse events received by the MAH during the PSUR review period were correctly coded (particularly in companies where listedness/expectedness is manually assigned to adverse events by the data entry specialist) and are appropriately presented in the line listings forming the PSUR source data and PSUR appendices. Any required corrections to the data within the MAH’s safety database need to be made at this stage, so that the PSUR author has ‘clean’ data available for the formal start of the PSUR writing process. The draft line listings should also be reviewed by the responsible Drug Safety Physician, to assess the totality and significance of safety data received by the MAH during the review period, in order to determine how any potential safety issues will be discussed/presented in the PSUR.
At Day -15, all key stakeholders in the PSUR preparation process (i.e. the Drug Safety Physician, Regulatory Affairs representative, and the Pharmacovigilance Officer) discuss all issues relating to the PSUR in question (e.g. new product registrations, marketing authorization withdrawals and relevant communications from the regulatory authorities (Regulatory Affairs), monitored safety topics (Drug Safety Physician), and any practical issues that may impact the availability of source data (the Pharmacovigilance Officer). This meeting should be organized and chaired by the responsible PV Medical Writer, who at the end of the meeting should have a complete picture of the PSUR data, any relevant safety and regulatory issues to be dealt with in the PSUR, and finally, the expected contribution from the team members.
5.2.5.2 Writing of the Draft PSUR
In general, formal writing activities commence in the first week after the DLP, as it is usually advisable to wait 3–4 days after the DLP before final line listings and summary tabulations are prepared. This is to ensure that sufficient time has been allowed for any case reports received on the actual DLP date to be entered into the MAH’s safety database and thus included in the PSUR data. Initiation of writing activities at this stage allows approximately 3 weeks for completion of the draft PSUR for preliminary review by the Drug Safety Physician.
5.2.5.3 Review of the Draft PSUR
The review process for PSURs will vary across companies, with some still using the traditional method of Word documents and comments by track changes. However, increasingly, companies are opting to use a Documentum-based electronic document management system or similar (tailored to each MAH’s needs) for review and storage of their PSURs.
5.2.5.4 QC Activities and PSUR Finalization
As a general rule, it is advisable to have the PSUR approved and signed off by the Qualified Person and/or Director responsible for safety by Day 55. This allows sufficient time for Regulatory Affairs to complete their in-house activities and submit the documents to the relevant authorities in a timely manner.
5.2.6 Generic Model of an EU PSUR
Each section of the EU PSUR is reviewed in this generic model, with a summary of the data to be presented in the section together with the key messages that should be highlighted.
| Period Covered by this Report | dd month year to dd month year |
| International Birth Date | dd month year |
| Data Lock Point | dd month year |
| Version, Date of Report | Final, dd month year |
| Abbreviations |
| Executive Summary |
| 1 Introduction |
| 2 Worldwide Marketing Authorization Status |
| 3 Update of Regulatory or Marketing Authorization Holder Actions taken for Reasons of Safety |
| 4 Changes to the Reference Safety Information |
| 5 Patient Exposure |
| 5.1 Post-marketing Exposure |
| 5.2 Clinical Trial Exposure |
| 6 Presentation of Individual Case Histories |
| 6.1 Scope of Presented Data and General Considerations |
| 6.2 Presentation of Line Listings |
| 6.3 Summary Tabulations |
| 6.4 Analysis of Individual Case Reports |
| 7 Studies |
| 7.1 Newly Analyzed Company-Sponsored Studies |
| 7.2 Targeted Safety Studies |
| 7.3 Published Safety Studies |
| 8 Other Information |
| 8.1 Efficacy-related Information |
| 8.2 Late-breaking Information |
| 8.3 Risk Management Plans |
| 8.4 Benefit-Risk Evaluation Reports |
| 9 Overall Safety Evaluation |
| 9.1 Changes in Listed Reactions |
| 9.2 Serious Unlisted Reactions |
| 9.3 Non-serious Unlisted Reactions |
| 9.4 Consumer Reports |
| 9.5 Drug Interactions |
| 9.6 Experience with Overdose, Deliberate or Accidental |
| 9.7 Drug Abuse and Misuse |
| 9.8 Medication Errors |
| 9.9 Experience with Pregnancy and Lactation |
| 9.10 Experience in Special Patient Groups |
| 9.11 Effects of Long-term Treatment |
| 10 Conclusions |
| 11 Appendices |
| Abbreviation | Definition |
| ADR | Adverse drug reaction |
| AE | Adverse event |
| CCSI | Company Core Safety Information |
| CIOMS | Council for International Organizations of Medical Sciences |
| DLP | Data Lock Point |
| EU | European Union |
| HCP | Healthcare Professional |
| IBD | International Birth Date |
| ICH | International Conference on Harmonisation |
| MAH | Marketing Authorization Application |
| MedDRA | Medical Dictionary for Regulatory Activities |
| PSUR | Periodic Safety Update Report |
| PT | Preferred term |
| RA | Regulatory Authority |
| RMP | Risk Management Plan |
| RSI | Reference Safety Information |
| SOC | System Organ Class |
| TTO | Time-to-onset |
| WWMA | Worldwide Marketing Authorization |
| Note: The table needs to be expanded and completed as required. | |
- introductory section stating the PSUR number, scope of presented data, the product’s history and approved indications;
- changes to marketing authorizations;
- regulatory or MAH actions taken for reasons of safety;
- changes to the Reference Safety Information (RSI);
- patient exposure;
- summary and number of cases presented in the PSUR;
- overall conclusion, with a summary any actions (if any) to be undertaken by the MAH.
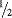 to
to 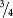 of a page) describe the product for which it is prepared (i.e. the active ingredient, therapeutic class, mechanism of action/pharmacology, approved indications, and recommended treatment doses).
of a page) describe the product for which it is prepared (i.e. the active ingredient, therapeutic class, mechanism of action/pharmacology, approved indications, and recommended treatment doses).- dates of marketing authorizations and renewals;
- any restrictions/qualifications on the marketing authorization that pertain to safety;
- approved indications and treatment populations;
- failed applications for marketing authorization (with rationale);
- MAH withdrawal of applications for marketing authorization (with rationale);
- withdrawal of marketing authorization;
- registered brand/trade names and launch dates.
- withdrawal or suspension of marketing authorizations;
- failure to obtain renewal of marketing authorization;
- any restrictions placed on product distribution;
- suspension/early termination of clinical studies;
- modification of the recommended treatment doses;
- changes in the target population and/or approved indications;
- changes in product formulation.
- number of units (tablets, packs, etc.) of product sold;
- number of prescriptions written.
| Product Formulation | Units Sold | Patient Exposure (in treatment-months or treatment-years) |
| Adult formulation | ||
| Paediatric formulation | ||
| Note: The calculation of patient exposure from the sales data needs to be explained in a footnote. | ||
- the study title/name/number;
- the number of planned patients;
- number of patients exposed to treatment during the review period;
- number of patients exposed to placebo or an active comparator;
- cumulative number of patient exposed to treatment.
- all serious and non-serious adverse events from all spontaneous sources (including healthcare professionals, regulatory authorities, the literature, and consumers);
- all serious adverse events from clinical trials, patient registries, and post-marketing studies that were considered as attributable to the drug by the investigator or the MAH, or for which a reporter causality was not provided.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


