Figure 3.1
Age at surgery according to the type of TAA
Actually, available literature about TAA is limited, and there has been very few medical studies in this group of patients for several reasons:
TAA is a rare disease.
TAA is usually not responsible for any symptoms before a complication occurs.
Discovery of TAA requires imaging of the aorta, usually indicated for other reasons.
The disease is usually discovered when the dilatation is large and therefore the period of time during which a therapy can be tested is limited.
Familial screening of TAA related for genetic defects should permit early discovery of some aortic diseases. However, recognition of the genetic nature of TAA and awareness of the cardiologic community are only recent.
In the absence of a large population of patients with long follow-up, it is difficult to test a medical therapy according to medical standards (i.e. evidence based medicine). The only studies available were conducted in selected populations with TAA of genetic aetiology, such as Marfan syndrome. This approach allows for early recognition of patients “at risk” of developing TAA (since they have a genetic defect), but Marfan syndrome remains a rare disease, and the studies performed therefore included limited number of patients (cf Marfan syndrome Chap. 4 for complete discussion). To date, no medical therapy has been clearly demonstrated to be associated with decreased mortality or complication rate.
Although aneurysms reaching a certain size are generally treated with surgery or endovascular therapy, many aspects of their medical management should be considered first.
Therefore, when caring for TAA patients medically, we lack evidenced based medicine, and are left with only reasoning, as was medicine in the preceding century, i.e., an art! As a result, the proposals made below cannot be considered as established truth, and the aim of this review is discuss the current state of the art.
If reasoning is the forefront on medical therapy in a patient with TAA, the first aim should be to assess the natural history of the disease and to seek for a modifiable etiologic factor, and treat it accordingly.
In fact TAA can be due to multiple etiologies, with varying prognosis (Fig. 3.1).
Aetiologies of TAA
From family studies, it is estimated that 20 % of TAAs are due to genetic diseases. The common pattern of inheritance seems to be autosomal dominant with different penetrance levels [2]. Among other risk factors, more important for the descending thoracic aorta, smoking has the strongest association with TAA. Dyslipidemia and hypertension are less powerful risk factors, considered to be associated mainly with the occurrence of AAA, although some data suggest that hypertension may actually be more closely associated with and is certainly a risk factor for dissection [3]. Men are more often affected than women (this is true whether an aneurysm is secondary to genetic factors or not). Advanced age, hypertension, chronic obstructive lung disease, and coronary artery disease are also associated risk factors for descending TAA [4].
Genetic Forms of TAA (Predominant in the Ascending Aorta)
The most common genetic aetiology for TAA is a mutation in the FBN1 gene, by far the most frequent cause of Marfan syndrome. Beyond FBN1 gene mutations, other genes have been implicated in the development of TAA, and can be grouped into different categories (Montalcino Aortic Consortium Classification) [5] (Table 3.1, Fig. 3.2).
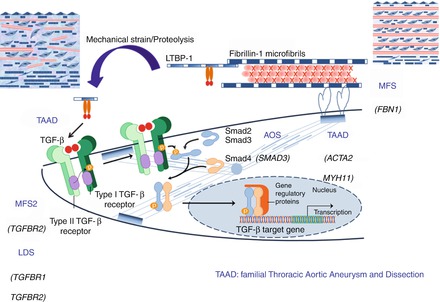
Table 3.1
Genetic etiologies of TAA (Montalcino Aortic Consortium)
Gene | Molecule | Phenotype |
|---|---|---|
ECM | ||
FBN1 | Fibrillin-1 | Marfan syndrome |
COL3A1 | Type3 procollagen | Vascular Ehlers–Danlos syndrome |
COL4A5 | Type4 procollagen | Alport syndrome |
EFEMP2 | Fibulin-4 | Cutis laxa |
TGFβ path | ||
TGFBR1 | TGF-β receptor-1 | FTAAD/LDS |
TGFBR2 | TGF-β receptor-2 | FTAAD/MFS/LDS |
TGFB2 | TGF-β2 | FTAAD |
SMAD3 | SMAD3 | FTAAD/AOS |
Contractile | ||
ACTA2 | α-actin | FTAAD |
MYH11 | Myosin heavy chain-11 | FTAAD |
MYLK | Myosin light chain kinase | FTAAD |
PRKG1 | cGMP-dependent | FTAAD |
FLNA | Filamin-A | Cerebral heterotopias/Aortic aneurysm |
TSC2 | Tuberin | Tuberous sclerosis complex |
Others | ||
JAG1 | JAGGED-1 | Alagille syndrome |
NOTCH1 | NOTCH-1 | Bicuspid aortic valve/Ao aneurysm |
SLC2A10 | Glucose transporter 10 | Arterial tortuosity syndrome |
Figure 3.2
More frequent genetic etiologies for TAAD (Thoracic Aortic Aneurysm Dissection); see text for abbreviations
1.
Mutation within genes coding for an extra-cellular matrix protein (FBN1, COL3A1). Such mutations are responsible for syndromic TAA, i.e. TAA associated with extra-aortic features (Marfan Syndrome, and vascular Ehlers Danlos syndrome).
(a)
Marfan syndrome is usually secondary to mutations in the FBN1 gene and leads to multisystem findings including skeletal features (tall stature, scoliosis, pectus deformities, elongated fingers and toes, hyperflexibility), ocular involvement (lens dislocation, high myopia), striae atrophiae, pneumothorax, and cardiovascular disease (aortic root aneurysm, aortic dissection, mitral valve prolapse) [6, 7]. See Marfan syndrome Chap. 4
(b)
Vascular Ehlers Danlos syndrome (vEDS) can be suspected on a variety of symptoms (medium sized arterial rupture, digestive complication such as bowel perforation, uterine rupture during pregnancy [8], and is confirmed nowadays by genetic testing (presence of a mutation at the COL3A1 gene encoding the pro-alpha 1 chain of type III procollagen. Actually, aortic aneurysms are not frequent in this pathology which is more responsible for aortic dissection than dilatation.
2.
Mutations within genes coding for a protein involved in the TGFB pathway (TGFB2, TGFBR1, TGFBR2, SMAD3), are associated with TAA but also aneurysms of other arteries, and possibly extra-aortic features (Aneurysm Osteoarthiritis Syndrome associated with SMAD3 mutation, LDS associated with mutation in TGFBR1 or TGFBR2, Marfan syndrome type II owing to TGFBR2 mutation) (Fig. 3.3).
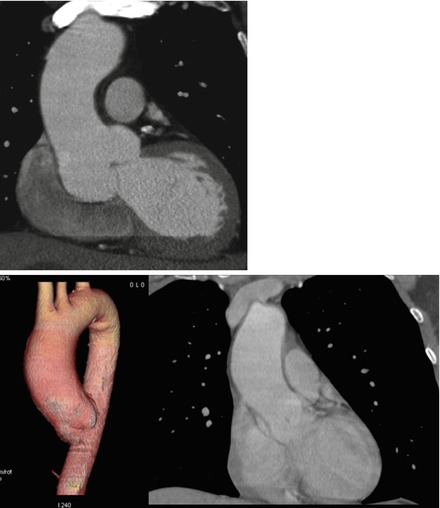
Figure 3.3
Top: aortic root dilatation in a patient with TGFB2 mutation: CT scanner. Maximal diameter at the level of the sinuses of Valsalva is 50 mm, and was considered as an indication for surgery. Bottom: aortic dilatation observed in a patient with a brother with BAV and TAA. Although the patient presented here did not show BAV, aortic dilatation of the tubular junction was shown on the CT scanner
(a)
Loeys-Dietz syndrome is a severe syndrome due to mutations in TGFBR1 and TGFBR2 genes and has a characteristic triad of craniofacial features (craniosynostosis, bifid uvula, hypertelorism), aortic root and branch vessel aneurysm and dissection, and arterial tortuosity [9, 10]. Mutations in TGFBR2 can also be responsible for familial forms of TAA with dominant autosomic inheritance pattern, and incomplete penetrance [11]. They can also be responsible for skeletal features similar to that observed in MFS syndrome related to FBN1 mutation [12].
(b)
Aneurysm Osteoarthritis Syndrome associates early-onset Osteoarthritis, Charcot-Marie-Tooth like neuropathy, autoimmune features, multiple arterial aneurysms and dissections [13] and secondary to SMAD3 mutations.
(c)
TGFB2 mutations are responsible for familial TAA with some skeletal features of Marfan Syndrome [14]
3.
Mutations within genes coding for components of the contractility apparatus of the smooth muscle cell (ACTA2, MYH11, PRKG1, FLNA).
(a)
ACTA2 gene mutations affect approximately 14 % of individuals with familial TAA disease in an serie from the USA but is much less frequent in our population. These mutations can be associated with livedo reticularis, iris flocculi, cerebral aneurysms, premature coronary and cerebrovascular disease, moyamoya [15, 16].
(b)
MYH11 gene mutations are responsible for familial forms of TAA often with patent ductus arteriosus [17]. They are very rare.
Dilatation of the aorta is mostly located at the level of the sinuses of Valsalva, and usually is symmetrical (Fig. 3.3). In some aetiologies (ACTA2, MYH11), dilatation can also be observed in the tubular portion of the ascending aorta.
Bicuspid Aortic Valve
Some TAA have been associated with a bicuspid aortic valve (BAV), consequently the frequency of TAA is greater in patients with BAV than in the general population. The reasons for this association are unclear, and 3 main hypothesis can be proposed [18]:
Aortic root dilatation may be constitutive, i.e. not reflecting a progressive increase in diameter with time. This may be close to the abnormal aortic cusps i.e. at the level of the sinuses of Valsalva, but not above the sino-tubular junction.
Aortic dilatation may be secondary to a primitive alteration in the aortic wall. In fact, the histological aspect of the aortic wall of TAA-operated patients with BAV is similar to that seen in patients with Marfan syndrome or other genetic forms of TAA.
Aortic dilatation results from alteration in the aortic flow pattern within the ascending aorta. The jet lesion historically proposed in patients with aortic stenosis has evolved into alterations in the normally laminar flow pattern of the ascending aorta, which is turbulent within the ascending aorta owing to anatomical abnormalities.
Maximum aortic diameter of TAA associated with BAV can either be localized at the level of the sinuses of Valsalva or above the sino-tubular junction. A relationship is observed between the type of the BAV and the anatomy of the aortic root: the antero-posterior diameter is increased when the BAV is related to a raphe between the two coronary artery cusps, type I R-L [19], and the aortic root progression rate is slow at the level of the sinuses of Valsalva [20]. In contrast, the dilatation of the aorta observed at the level of the tubular aorta is independent of the type of BAV and the dilatation rate is greater, which suggests that it may reflect alterations in the aortic wall and require specific care [20]. Indeed, when the dilatation rate of a population with BAV is studied, two groups are observed: patients who do not dilate over time, and those who do (Fig. 3.4).
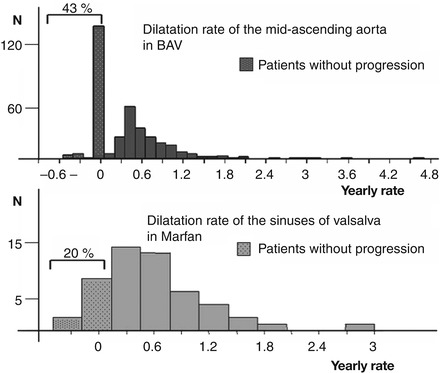
BAV can be associated with TAA and coarctation of the aorta. Familial forms exist, but with variability (one family member present aortic dilatation whereas a second may present BAV and a third aortic dilatation and another BAV without aortic dilatation). This requires familial screening to be systematic when a BAV is observed [21]. However, no genetic defect has been clearly associated with BAV besides NOTCH1 mutation very seldom.
Turner
Women with Turner syndrome in whom aortic dilatation can also be associated with BAV and coarctation of the aorta form a specific subgroup. In these small-statured women normalization of aortic diameter is essential since the absolute diameter may under-evaluate the aortic dilatation [23]. Aortic dilatation in patients with Turner syndrome has been reported to occur in up to 40 % of cases. In this population, special care should be taken in cases of pregnancy, which is generally considered to be contraindicated in women with aortic dilatation, BAV and/or coarctation [24]. Complete imaging of the aorta (MRI or CT scanner) are necessary in these women. Patients with an index >2 cm/m2 in the ascending aorta should be followed yearly as the risk for aortic dissection increases.
Others
Other aneurysms are neither related to any known mutation nor associated with BAV, and can be observed as sporadic or familial diseases. When familial, they are usually transmitted with a autosomal dominant pattern, and intensive research is ongoing to identify new genes involved. When sporadic, which is by far the more frequent form, the predisposing factors are usually haemodynamic (hypertension), and patient age is usually around 70.
Some specific aetiologies are seldom responsible for TAA, and are usually found in specific context: these are inflammatory diseases (see Chap. 2) (Giant cell arteritis, Takayasu disease, Kawasaki disease, Behcet Syndrome, aortitis associated with HLAB27…), or infectious diseases (Syphilis, Aspergillus, bacterial aortitis…). Obviously, when an active process responsible for aortic dilatation (such as infection) is ongoing, its treatment is warranted; however, aortic dilatation is sometimes observed years after the inflammatory phase of the disease, at a time when all therapy has long been stopped.
Descending Aorta Aneurysms
In contrast to aneurysms of the ascending aorta, aneurysms of the descending aorta are similar to aneurysms of the abdominal aorta from an epidemiological and pathophysiological point of view. Familial aggregation is present but genetic transmission is unproven and association with risk factors for atherosclerosis is the rule. In these cases, aortic dilatation is usually diffuse and not limited to a specific section. Calcification of the aortic wall is often observed. In these patients, it is obviously important to limit the risk factors.
A specific situation is the dilatation that can be observed at the initial part of the descending aorta, just below left subclavian artery level, often observed in patients with BAV and/or aortic coarctation. This dilatation appears to be present very early in life and its significance is unclear, although of the descending aorta dissections have been reported in patients with BAV [25].
Pathophysiology
TAA of the Ascending Aorta
Ascending aorta pathologies are chronic aneurismal diseases, corresponding to a progressive dilatation of the aorta, leading finally to rupture, and/or acute dissection corresponding to an intra-parietal rupture. These two pathologies are related to a progressive (aneurysms) or acute (dissections) degradation of the insoluble extracellular matrix proteins of the arterial wall, mainly elastin and collagens, which give the solidity to the arterial wall. This degradation is the fact of specific medial areas of mucoid degeneration (also formerly misnamed cystic medial necrosis), characterised by the local enrichment of alcianophilic glycosaminoglycans, vacuoles corresponding to the disappearance of smooth muscle cells, and local degradation of extracellular proteins, including disorganised adhesive proteins such as fibronectin and fibrillin and the rupture of insoluble elastin and collagen. The pathology of aneurysm and/or dissection does not differ in genetic or non-genetic aetiologies. They only clinically appear in younger patients as degenerative forms.
Role of Proteases
Fibrillin is not directly a component of the insoluble extracellular matrix of the arterial wall. Elastin and collagens are the main insoluble and hydrophobic components of the wall, giving it a strong support for resisting blood pressure (elementary contention function). Elastin is involved in wall elasticity, and is the main structural component of resistance to dilatation. Collagen is the main structural component of resistance to rupture. Both pathologies are linked to the proteolytic degradation of elastin (aneurysm) and collagen (dissection and rupture). Therefore, there is a tremendous interest in defining the panel of proteases involved in extracellular matrix degradation in Marfan syndrome and related diseases. The abundance and activity of Matrix metalloproteinases (MMPs) have been shown to be related to TAA formation in numerous studies [26]. Matrix metalloproteinase-2 is produced in mesenchymal cells; MMP-9 is produced in macrophages. We have identified the MMP-7 (matrilysin) and MMP-3 (stromelysin) as MMPs preferentially localised within the areas of mucoid degeneration [27], which probably results from their particular affinity for sulfated glycosaminoglycans.
In contrast, the data concerning serine protease activities present in aneurysm are scarce. We reported the presence of thrombin within the areas of mucoid degeneration, due, once again, to its affinity for glysosaminoglycans [28]. Interestingly enough, we recently explored the role of the plasminergic system in aneurysms of the ascending aorta, including Marfan [29]. Beside the activation of plasminogen after its binding to fibrin (fibrinolytic system), plasminogen could be activated by the plasminogen activators (PAs) expressed by mesenchymal cells. Activated plasmin released by the interaction between plasminogen and cell-derived PAs, catalysed by membrane proteins, lead to fibronectin degradation, cell detachment [30] and apoptosis [31]. On the other hand, plasmin is able to activate MMPs, to degrade adhesive fibronectin and fibrillin, etc. and therefore to provoke the release and activation of TGF-beta of its matrix storage sites. We reported that plasminogen is transferred better from plasma to an aneurismal wall than to a normal aortic wall, that t-PA and u-PA are more expressed in an aneurismal wall than in a normal one, and therefore that generation of plasmin is enhanced in aneurismal walls as compared to normal walls, leading to an increase in TGF-beta bioavailability [29] in aneurysm of the ascending aorta. Since plasmin generation could participate to cell disappearance, MMP activation, and TGF-beta release, the fibrinolytic system is probably an important target for preventing dilatation [32]. In parallel, plasmin is also involved in dissecting pathology. In particular circulating plasmin-antiplasmin complex and fibrin degradation product have been proposed as markers of acute dissection, but this is probably due to the fibrinolysis of the clot in the false channel [33]. Nevertheless the participation of tissue plasmin is not excluded.
Role of TGF-Beta
In a mouse model KI for a FBN1 mutation [34], aortic dilatation occurs in heterozygous mice, and increased P-Smad 2 (the intracellular effecter for TGFB2) was observed in the aortic wall of pathologic mice compared to normal mice [34]. TGF-β neutralising antibodies were able to decrease the level of P-Smad 2 within the aortic wall. Besides, blocking the TGF-β pathway by the use of specific antibodies prevented abnormal aortic dilatation in this model. The idea has therefore emerged that the increased TGF-β pathway was responsible for the main anomalies associated with Marfan syndrome, rather than an abnormal structural protein directly leading to weakening of the extracellular matrix and tissues.
However, TGF-beta 1 activates both Smad and non-Smad pathways, and we were not able to find any activation of these non-Smad pathways in the aortic wall of patients with Marfan syndrome, nor were we able to find increased mRNA levels for TGF-beta 1 within the aortic wall [35]. This was true for the aortic walls of patients with Marfan syndrome as well as for aortic walls from patients with aortic aneurysms from other aetiologies (bicuspid aortic valve, non syndromic TAA).
In contrast, we were able to demonstrate an increase in P-Smad 2 in smooth muscle cells from the aortic wall of patients with Marfan syndrome but also patients with thoracic aortic aneurysms from other aetiologies [35]. Actually, we and others have also reported increased P-Smad-2 in the aortic wall of patients with TAA secondary to mutation in the TGF-β receptor, despite the fact that the mutation in the TGF-β receptor alters (blocks) the transmission of the signal [9, 36]; this suggests that increased Psmad-2 within the aortic wall is not secondary to increased TGF-β activation in human aorta. Beside, no clear association in localisation could be found between the Smad 2 nuclear levels and the TGF-β extracellular staining in the aortic wall of patients with aneurysmal aorta, also suggesting the absence of a direct link between the two observations [35].
All this data questions the simple cause and effect relationship that has been proposed between TGF-β activation and aortic root dilatation in Marfan syndrome, but widens the potential importance of the TGF-β pathway alteration in the aortic aneurysm disease: it suggests that actually, increased Smad-2 within the aortic wall is related to a “common pathway” observed in all forms of TAA, which could either be responsible for (as suggested by the beneficial effect of neutralising antibodies) or responsive to (as would have been anticipated by the known pro-fibrotic and anti-proteolytic effects of TGF-β) the dilatation of the aorta. This last hypothesis is also compatible with the correlation observed between increased Smad-2 level and the degree of elastic fibre fragmentation that we observed in aortic aneurismal wall of diverse aetiologies [35].
Dissociation between TGF-β activation and increase in Smad-2 signalling was further supported by recent experiments from our group. We were able to demonstrate that (1) increased P-Smad-2 was specific to smooth muscle cell (SMC) i.e. not present in fibroblasts obtained from the aortic wall despite the fact that all cells should be submitted to the same TGF-β stimulation coming from the extracellular matrix within the same aortic wall, (2) increased P-Smad-2 was associated with increased Smad-2 RNA level within SMC (which was not present in the fibroblasts coming from the same aortic wall) (3) this deregulation of the Smad 2 pathway within the SMC was heritable, i.e. increased P-Smad 2 concentration was maintained during SMC culture, despite the absence of TGF-β within the culture milieu [37], indicating an epigenetic control of increased Smad-2 within the SMC. Actually this epigenetic control was further suggested by chromatin immuno-precipitation, showing alterations of the histones linked to the promoter of the Smad-2 gene within the SMC [37]. These observations were made in cells derived from aneurismal aortic wall from various aetiologies (i.e. patients with Marfan syndrome but also patients with aortic aneurysms from other aetiologies) compared to SMC derived from normal human aorta.
As a conclusion regarding these observations, we can say that increased P-Smad-2 within the SMC of aortic aneurismal wall is observed whatever the aetiology of the aneurysm, and that its relation to TGF-β activation is not clearly established. Actually, this may be a compensatory mechanism induced within the smooth muscle cell of aneurismal wall independent from the aetiology of the aneurysm.
This discussion is of importance the interpretation of the beneficial effect of losartan if proven, because this mechanism should determine which is the population that could benefit from this therapy: only Marfan patients or all patients with TAA (see below for discussion)
Imaging Follow-up and Risk of Complications
A patient with a TAA should be followed-up by a cardiologist, and have regular imaging of the aorta. It is usually recommended that TTE should be repeated after 6 months to ensure the absence of rapid evolution of the aortic dilatation when a TAA is recognized for the first time in a patient [21, 38]. Thereafter a yearly imaging is considered sufficient, unless the aortic diameter is coming close to the surgical threshold. Then echocardiography every 6 months may be wise [38]. Diameter expansion, severity of aortic regurgitation, and left ventricular function may be correctly evaluated when the echocardiographic window is adequate.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


