Chapter 11 Pharmacology
Its role in drug discovery
Introduction
• Selectivity screening, consisting of in vitro tests on a broad range of possible drug targets to determine whether the compound is sufficiently selective for the chosen target to merit further investigation
• Pharmacological profiling, aimed at evaluating in isolated tissues or normal animals the range of effects of the test compound that might be relevant in the clinical situation. Some authorities distinguish between primary pharmacodynamic studies, concerning effects related to the selected therapeutic target (i.e. therapeutically relevant effects), and secondary pharmacodynamic studies, on effects not related to the target (i.e. side effects). At the laboratory level the two are often not clearly distinguishable, and the borderline between secondary pharmacodynamic and safety pharmacology studies (see below) is also uncertain. Nevertheless, for the purposes of formal documentation, the distinction may be useful
• Testing in animal models of disease to determine whether the compound is likely to produce therapeutic benefit
• Safety pharmacology, consisting of a series of standardized animal tests aimed at revealing undesirable side effects, which may be unrelated to the primary action of the drug. This topic is discussed in Chapter 15.
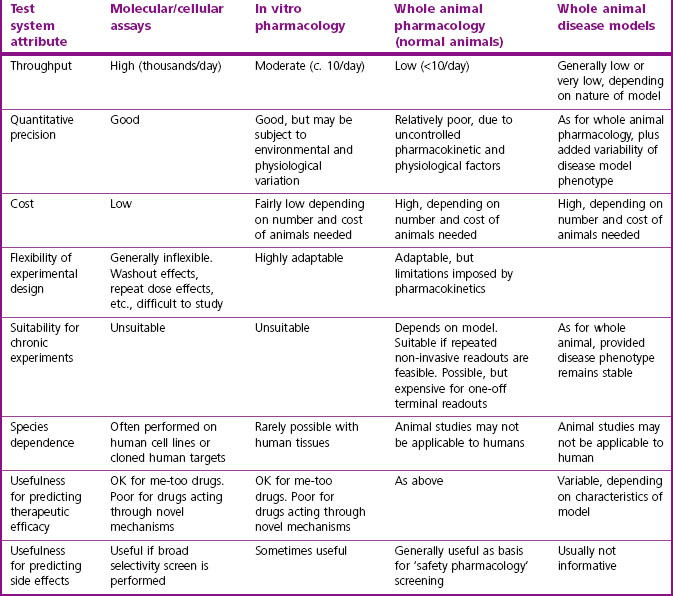
The strengths and weaknesses of these test systems are summarized in Table 11.1.
Pharmacological characterization of a candidate compound often has to take into account active metabolites, based on information from drug metabolism and pharmacokinetics (DMPK) studies (see Chapter 10). If a major active metabolite is identified, it will be necessary to synthesize and test it in the same way as the parent compound in order to determine which effects (both wanted and unwanted) relate to each. Particular problems may arise if the metabolic fate of the compound shows marked species differences, making it difficult to predict from animal studies what will happen in humans.
Screening for selectivity
The selectivity of a compound for the chosen molecular target needs to be assessed at an early stage. Compounds selected for their potency, for example on a given amine receptor, protease, kinase, transporter or ion channel, are very likely to bind also to related – or even unrelated – molecular targets, and thereby cause unwanted side effects. Selectivity is, therefore, as important as potency in choosing potential development candidates, and a ‘selectivity screen’ is usually included early in the project. The range of targets included in such a screen depends very much on the type of compound and the intended clinical indication. Ligands for monoamine receptors and transporters form a large and important group of drugs, and several contract research organizations (e.g. CEREP, MDL) offer a battery of assays – mainly binding assays, but also a range of functional assays – designed to detect affinity for a wide range of receptors, transporters and channels. In the field of monoamine receptors, for example, it is usually important to avoid compounds that block or activate peripheral muscarinic receptors, adrenergic receptors or histamine (particularly H1) receptors, because of the side effects that are associated with these actions, and a standard selectivity test battery allows such problems to be discovered early. Recently, several psychotropic and anti-infective drugs have been withdrawn because of sudden cardiac deaths, probably associated with their ability to block a particular type of potassium channel (known as the hERG channel; see Chapter 16) in myocardial cells. This activity can be detected by electrophysiological measurements on isolated myocardial cells, and such a test is now usually performed at an early stage of development of drugs of the classes implicated in this type of adverse reaction.
Interpretation of binding assays
Binding assays, generally with membrane preparations made from intact tissues or receptor-expressing cell lines, are widely used in drug discovery projects because of their simplicity and ease of automation. Detailed technical manuals describing the methods used for performing and analysing drug binding experiments are available (Keen, 1999; Vogel, 2002). Generally, the aim of the assay is to determine the dissociation constant, KD, of the test compound, as a measure of its affinity for the receptor. In most cases, the assay (often called a displacement assay) measures the ability of the test compound to inhibit the binding of a high-affinity radioligand which combines selectively with the receptor in question, correction being made for ‘non-specific’ binding of the radioligand.
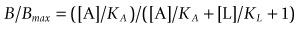 (1)
(1)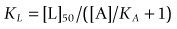 (2)
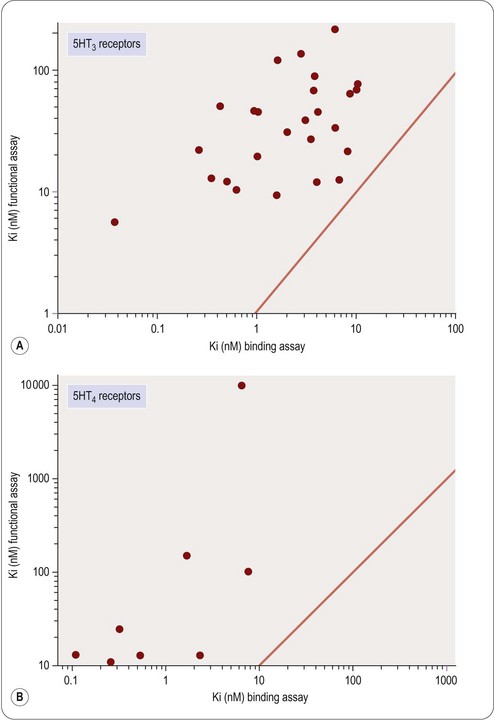
(2)This is often known as the Cheng–Prusoff equation, and is widely used to calculate KL when [L]50, [A] and KA are known. It is important to realize that the Cheng–Prusoff equation applies only (a) at equilibrium, (b) when the interaction between A and L is strictly competitive, and (c) when neither ligand binds cooperatively. However, an [L]50 value can be measured for any test compound that inhibits the binding of the radioligand by whatever mechanism, irrespective of whether equilibrium has been reached. Applying the Cheng–Prusoff equation if these conditions are not met can yield estimates of KL that are quite meaningless, and so it should strictly be used only if the conditions have been shown experimentally to be satisfied – a fairly laborious process. Nevertheless, Cheng–Prusoff estimates of ligand affinity constants are often quoted without such checks having been performed. In most cases it would be more satisfactory to use the experimentally determined [L]50 value as an operational measure of potency. A further important caveat that applies to binding studies is that they are often performed under conditions of low ionic strength, in which the sodium and calcium concentrations are much lower than the physiological range. This is done for technical reasons, as low [Na+] commonly increases both the affinity and the Bmax of the radioligand, and omitting [Ca2+] avoids clumping of the membrane fragments. Partly for this reason, ligand affinities estimated from binding studies are often considerably higher than estimates obtained from functional assays (Hall, 1992), although the effect is not consistent, presumably because ionic bonding, which will be favoured by the low ionic strength medium, contributes unequally to the binding of different ligands. Consequently, the correlation between data from binding assays and functional assays is often rather poor (see below). Figure 11.1 shows data obtained independently on 5HT3 and 5HT4 receptors; in both cases the estimated KD values for binding are on average about 10 times lower than estimates from functional assays, and the correlation is very poor.
Pharmacological profiling
• Ligand-binding assay on membrane fragments from a cell line expressing the cloned receptor
• Inhibition of agonist activity in a cell line, based on a functional readout (e.g. raised intracellular calcium)
• Antagonism of a selective agonist in an isolated tissue (e.g. smooth muscle, cardiac muscle). Such assays will normally be performed with non-human tissue, and so interspecies differences in the receptor need to be taken into account. Sometimes specific questions have to be asked about effects on human tissues for particular compounds and then collecting viable tissues to use becomes a major challenge
• Antagonism of the response (e.g. bronchoconstriction, vasoconstriction, increased heart rate) to a selective receptor agonist in vivo. Prior knowledge about species specificity of the agonist and antagonist is important at this stage.
• Do the molecular and cellular effects measured in screening assays actually give rise to the predicted pharmacological effects in intact tissues and whole animals?
• Does the compound produce effects in intact tissues or whole animals not associated with actions on its principal molecular target?
• Is there correspondence between the potency of the compound at the molecular level, the tissue level and the whole animal level?
• Do the in vivo potency and duration of action match up with the pharmacokinetic properties of the compound?
• What happens if the drug is given continuously or repeatedly to an animal over the course of days or weeks? Does it lose its effectiveness, or reveal effects not seen with acute administration? Is there any kind of ‘rebound’ after effect when it is stopped?
In vitro profiling
Measurements on isolated tissues
Studies on isolated tissues have been a mainstay of pharmacological methodology ever since the introduction of the isolated organ bath by Magnus early in the 20th century. The technique is extremely versatile and applicable to studies on smooth muscle (e.g. gastrointestinal tract, airways, blood vessels, urinary tract, uterus, biliary tract, etc.) as well as cardiac and striated muscle, secretory epithelia, endocrine glands, brain slices, liver slices, and many other functional systems. In most cases the tissue is removed from a freshly killed or anaesthetized animal and suspended in a chamber containing warmed oxygenated physiological salt solution. With smooth muscle preparations the readout is usually mechanical (i.e. tension, recorded with a simple strain gauge). For other types of preparation, various electrophysiological or biochemical readouts are often used. Vogel (2002) and Enna et al. (2003) give details of a comprehensive range of standard pharmacological assay methods, including technical instructions.
Studies of this kind have the advantage that they are performed on intact normal tissues, as distinct from isolated enzymes or other proteins. The recognition molecules, signal transduction machinery and the mechanical or biochemical readout are assumed to be a reasonable approximation to the normal functioning of the tissue. There is abundant evidence to show that tissue responses to GPCR activation, for example, depend on many factors, including the level of expression of the receptor, the type and abundance of the G proteins present in the cell, the presence of associated proteins such as receptor activity-modifying proteins (RAMPs; see Morfis et al., 2003), the state of phosphorylation of various constituent proteins in the signal transduction cascade, and so on. For compounds acting on intracellular targets, functional activity depends on permeation through the membrane, as well as affinity for the target. For these reasons – and probably also for others that are not understood – the results of assays on isolated tissues often differ significantly from results found with primary screening assays. The discrepancy may simply be a quantitative one, such that the potency of the ligand does not agree in the two systems, or it may be more basic. For example, the pharmacological efficacy of a receptor ligand, i.e. the property that determines whether it is a full agonist, a partial agonist, or an antagonist, often depends on the type of assay used (Kenakin, 1999), and this may have an important bearing on the selection of possible development compounds. Examples that illustrate the poor correlation that may exist between measurements of target affinity in cell-free assay systems, and functional activity in intact cell systems, are shown in Figures 11.1 and 11.2. Figure 11.1 shows the relationship between binding and functional assay data for 5HT3 and 5HT4 receptor antagonists. In both cases, binding assays overestimate the potency in functional assays by a factor of about 10 (see above), but more importantly, the correlation is poor, despite the fact that the receptors are extracellular, and so membrane penetration is not a factor. Figure 11.2 shows data on tyrosine kinase inhibitors, in which activity against the isolated enzyme is plotted against inhibition of tyrosine phosphorylation in intact cells, and inhibition of cell proliferation for a large series of compounds. Differences in membrane penetration can account for part of the discrepancy between enzyme and cell-based data, but the correlation between intracellular kinase inhibition and blocking of cell proliferation is also weak, which must reflect other factors.
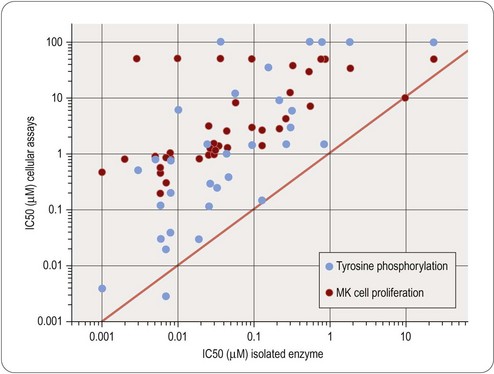
Fig. 11.2 Correlation of cellular activity of EGFR receptor kinase inhibitors with enzyme inhibition.
Data from Traxler et al., 1997.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


