Which drug was taken with the aspirin and inhibited its aggregatory effects in response to ADP or collagen?
a. Acetaminophen
b. Clopidogrel
c. Dabigatran
d. Ibuprofen
e. Warfarin
192. A 57-year-old patient complains of muscle aches, pain, and tenderness. These affect the legs and trunk. There is no fever, bruising, or any recent history of muscle trauma or strains (as from excessive exercise). He has myoglobinuria, a clinically significant fall of creatinine clearance, and a rise of plasma creatine kinase (CK) to levels nearly ten times the upper limit of normal. Which drug is the most likely cause of these findings?
a. Aspirin (low dose) for its cardioprotective/antiplatelet effects
b. Captopril for hypertension and heart failure
c. Carvedilol for hypertension, heart failure, and angina prophylaxis
d. Furosemide as adjunctive management of his heart failure
e. Rosuvastatin to control his hypercholesterolemia and the associated risks
193. A coronary artery sample was removed from a healthy animal, put inside a suitable oxygenated salt and nutrient solution, and connected to a transducer that measured increases (contraction) or decreases (relaxation) of smooth muscle tension (force). ACh was then added to give the cumulative concentrations shown below.
As expected, in the control setting (left) ACh caused concentration-dependent vasorelaxation (equivalent to vasodilation in the intact animal). The ACh was washed out several times, control conditions returned. The conditions were manipulated, and then the ACh dose-response experiment was repeated (right). Now the data show increased tension developed by the muscle sample (vasoconstriction in vivo) in response to ACh and incremental increases of its concentration.
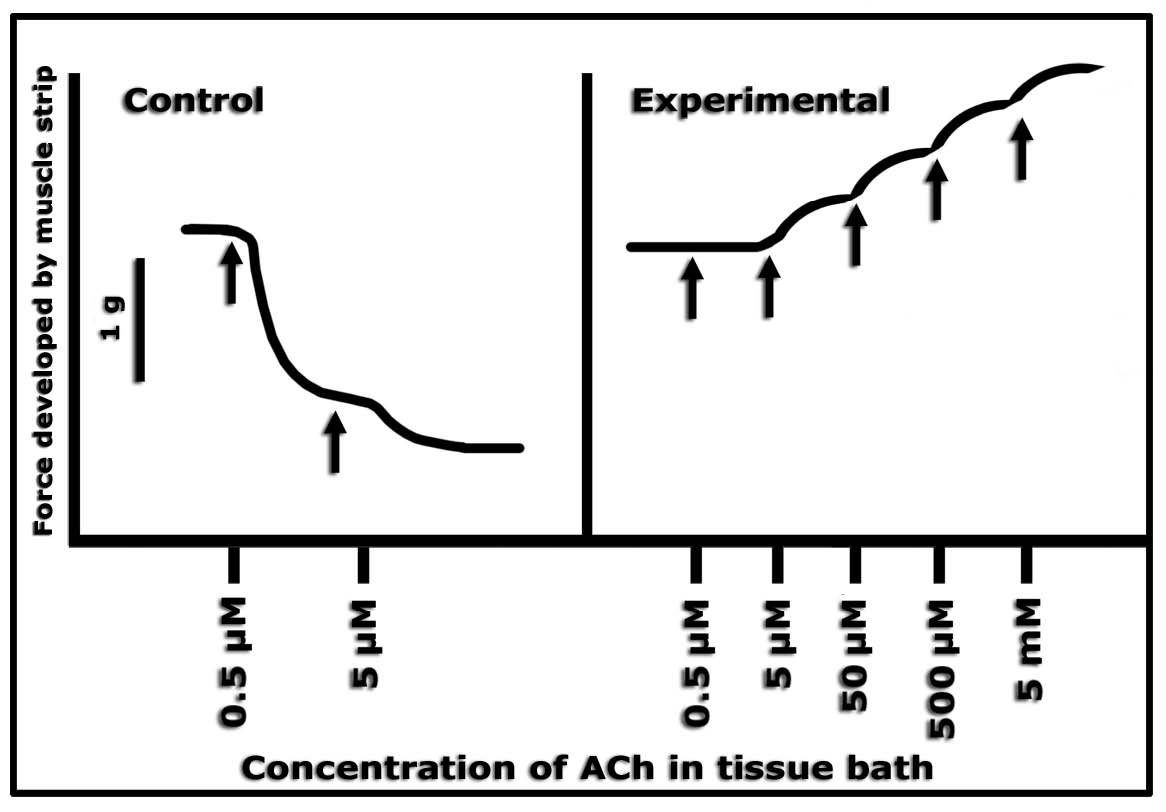
Which one of the following summarizes what was most likely done to the vessel under the experimental conditions, before retesting the responses to added ACh? Assume that the vascular responses in this animal model are identical to those that would occur in a human.
a. Endothelium was removed mechanically
b. Isoproterenol was added right before ACh
c. Muscarinic receptors were blocked with atropine
d. Sample was pretreated with prazosin
e. Tissue was pretreated with botulinum toxin
194. A 28-year-old woman is receiving drug therapy for essential (primary) hypertension. She subsequently becomes pregnant. You realize that the drug she’s been taking for her high blood pressure can have serious, if not fatal, effects on the fetus (it is in pregnancy category X). As a result, you stop the current antihypertensive drug and substitute another that is deemed to be equiefficacious in terms of her blood pressure, and safer for the fetus. Which drug was she most likely taking before she became pregnant?
a. α-Methyldopa
b. Captopril
c. Furosemide
d. Labetalol
e. Verapamil
195. We treat a patient with a drug that affects the clotting-thrombolytic systems for a time sufficient to let the drug’s effects and blood levels stabilize at a therapeutic level. We then isolate platelets from a blood sample and test their in vitro aggregatory responses to ADP, collagen, PAF, and thromboxane A2. Aggregatory responses to ADP are inhibited; responses to the other platelet proaggregatory agonists are unaffected. Which drug exhibits these properties?
a. Aspirin
b. Bivalirudin
c. Clopidogrel
d. Heparin
e. Warfarin
196. Your newly diagnosed hypertensive patient has vasospastic angina. Which drug or drug class would be the most rational for starting anti-hypertensive therapy because it exerts not only antihypertensive effects, but also directly lowers myocardial oxygen demand and consumption and tends to inhibit cellular processes that otherwise favor coronary vaso-spasm? Assume there are no other specific contraindications to the drug you choose.
a. Angiotensin-converting enzyme (ACE) inhibitor or angiotensin receptor blocker
b. β-Adrenergic blocker
c. Nifedipine
d. Thiazide diuretic
e. Verapamil (or diltiazem)
197. You are reviewing the medication history of a 59-year-old man. He has been taking ramipril and pravastatin for the last 5 years. Other medications include metformin for type 2 diabetes mellitus, and escitalopram to help manage his depression. At his last clinic visit, a year ago, he was told to continue his current medications but he was also started on slow-release niacin because diet, exercise and other lifestyle modifications, and his current medications were not adequate. What was the most likely reason for adding the niacin?
a. Counteract deficiencies of B-vitamin absorption caused by the antidepressant
b. Counteract polyphagia, and over-eating, caused by the metformin.
c. Lower HDL and triglyceride levels that did not respond adequately to the statin
d. Prevent statin-induced neuropathy
e. Slow the progression of diabetic nephropathy caused by the ACE inhibitor
198. Quinidine is ordered for a patient with recurrent atrial fibrillation and who refuses any interventions other than drugs in an attempt to terminate and control the arrhythmia. He has some pulmonary fibrosis and a thyroid disorder—both leading you to conclude that amiodarone therapy might not be the best approach. Which statement applies to the quinidine?
a. Decreases SA nodal automaticity due to a strong anticholinergic/vagolytic effect
b. Is likely to increase blood pressure via a direct vasoconstrictor effect
c. Is contraindicated if the patient also requires anticoagulant therapy
d. Tends to increase electrical impulse conduction velocity through the AV node
e. Will increase cardiac contractility (positive inotropic effect) independent of its antiarrhythmic effects
199. A woman has received a cardiovascular drug that is absolutely contraindicated (category X) in pregnancy, but in the absence of pregnancy would have been deemed beneficial because of a particular cardiovascular disorder she has. Unfortunately, neither she nor her physician (whom she rarely visited) knew she was pregnant until she had just started the second trimester. The drug is stopped (and a suitable and safer alternative is started), but it is too late. Her baby is delivered prematurely, and stillborn. It is obvious from examination of the baby that there is a nasal deformity: the nose is flattened into the face, with no apparent bridge. Standard x-rays reveal stippling of all the epiphyses. These responses are characteristic of which drug?
a. Clonidine
b. Heparin, low molecular weight (eg, enoxaparin)
c. Hydrochlorothiazide
d. Nitroglycerin
e. Warfarin
200. A patient who has been taking an oral antihypertensive drug for about a year develops a positive Coombs’ test, and now you are worried about the possibility (although low) that hemolytic anemia may develop if the drug is continued. Which drug was the most likely cause?
a. Captopril
b. Clonidine
c. Labetalol
d. Methyldopa
e. Prazosin
201. A patient presents with severe hypertension and tachycardia. Blood chemistry results, radiologic studies, and the overall clinical presentation point to pheochromocytoma. The tumor appears operable, but the patient will have to wait a couple of weeks for the adrenalectomy. We prescribe phenoxybenzamine in the interim, with the goal of suppressing some of the major signs and symptoms caused by the tumor and the massive amounts of epinephrine it is releasing. Which of the following best summarizes what phenoxybenzamine does, or how it acts?
a. Controls blood pressure by blocking α-adrenergic receptors in the peripheral vasculature
b. Controls heart rate by selectively blocking β1-adrenergic receptors
c. Inhibits catecholamine synthesis in the adrenal (suprarenal) medulla
d. Lowers blood pressure by inhibiting angiotensin converting enzyme
e. Stimulates catechol-O-methyltransferase, thereby facilitating epinephrine’s metabolic inactivation
202. A 30-year-old man who has a history of asthma has just been diagnosed Stage 1 essential hypertension. He regularly uses an inhaled corticosteroid, which seems to work well as a control medication, but also needs to use an albuterol inhaler about once every 3 weeks for suppression of asthma attacks (rescue therapy). Which antihypertensive drug or drug class poses the greatest risk of exacerbating the patient’s asthma and counteracting the desired pulmonary effects of the albuterol, even though it might control his blood pressure well?
a. Diltiazem
b. Hydrochlorothiazide
c. Labetalol
d. Ramipril
e. Verapamil
203. Digoxin affects a host of cardiac electrophysiologic properties. Some of its effects are caused directly by the drug. Others are indirect: they may involve increasing “vagal tone” to the heart or other compensations that arise when cardiac output is improved in a patient with heart failure. For some parameters the direct and indirect effects may be qualitatively (but not quantitatively) opposing, but one will predominate over the other. What is an expected and usually predominant effect of the drug?
a. Increased rate of SA nodal depolarization
b. Reduced atrial automaticity
c. Reduced ventricular automaticity
d. Slowed AV nodal conduction velocity
e. Slowed conduction velocity through the atrial myocardium and His-Purkinje system
204. A patient has Stage 2 essential hypertension and heart failure. After evaluating the responses to many other antihypertensive drugs, alone and in combination, the physician concludes that it would be reasonable to try hydralazine. Which drug(s) is/are likely to be needed, as add-ons (adjuncts), to manage the expected and unwanted cardiovascular and renal side effects of the hydralazine?
a. Captopril plus nifedipine
b. Digoxin plus spironolactone
c. Digoxin plus vitamin K
d. Hydrochlorothiazide and a β-blocker
e. Nitroglycerin
f. Triamterene plus amiloride
205. A healthy adult subject participating in a clinical trial is given an intravenous injection of a test drug. Both blood pressure and total peripheral resistance rise promptly. This is followed immediately by a reduction of heart rate. In repeated experiments we find that the vasopressor response is not affected by pretreatment with prazosin. However, pretreatment with atropine prevents the cardiac chronotropic response. The test drug was most likely which of the following?
a. Angiotensin II
b. Dobutamine
c. Isoproterenol
d. Norepinephrine
e. Phenylephrine
206. We are administering nitroprusside intravenously for control of severe hypertension during surgery. The dose has gotten too high, and the drug has been administered too long. Refractoriness to the antihypertensive effects has occurred. Blood pressure is rising, and other signs and symptoms of potentially severe toxicity develop. What nitroprusside metabolite accounts for or at least contributes to these problems?
a. A highly efficacious α-adrenergic agonist
b. An extraordinarily potent and irreversible Na-K-ATPase inhibitor
c. An irreversible antagonist for angiotensin at the A-II receptors
d. Cyanide
e. Nitric oxide
207. A 66-year-old man who lives in a small rural town, and who has been treated by his family doctor for decades, presents at your medical center. He has coronary atherosclerosis and “mild” heart failure that has been treated for the last 10 years with digoxin and several other drugs. His chief complaints are nausea, vomiting, and diarrhea, which have not resolved despite a recommendation from his physician to take prescription medications for those conditions. His ECG reveals a bigeminal rhythm and second-degree heart block. A drug-drug interaction is suspected. What coadministered drug most likely provoked the problem?
a. Captopril
b. Cholestyramine
c. Furosemide
d. Lovastatin
e. Nitroglycerin
208. We have a patient who is diagnosed with variant (vasospastic) angina. Which drug would be most appropriate, and generally regarded as most effective, for long-term therapy aimed at reducing the incidence or severity of the coronary vasospasm?
a. Aspirin
b. Atorvastatin
c. Diltiazem
d. Nitroglycerin
e. Propranolol
209. A 56-year-old man has heart failure. His family doctor, who has been treating him since he was a young lad, has been treating him with digoxin, furosemide, and triamterene for several years. The patient now develops atrial fibrillation, and so his doctor starts quinidine and clopidogrel. What is the most likely outcome of adding the quinidine?
a. Development of signs and symptoms of quinidine toxicity (cinchonism)
b. Hyponatremia due to quinidine’s ability to enhance diuretic-induced sodium loss
c. Onset of signs and symptoms of digoxin toxicity
d. Precipitous development of hypokalemia
e. Prompt suppression of cardiac contractility, onset of acute heart failure
210. Flecainide and propafenone are in Vaughan-Williams (antiarrhythmic) Class I-C. What is the clinically relevant “take home” message about this class of drugs?
a. Are only given for arrhythmias during acute myocardial infarction
b. Are particularly suited for patients with low ejection fractions or cardiac output
c. Are preferred drugs (drugs of choice) for relatively innocuous ventricular arrhythmias
d. Cause pulmonary fibrosis and a hypothyroid-like syndrome when given long term
e. Have a significant pro-arrhythmic effect (induction of lethal arrhythmias)
211. You want to compare and contrast the cardiac and hemodynamic profiles of immediate-acting dihydropyridine-type calcium channel blockers (CCBs) and the nondihydropyridine, verapamil (or diltiazem). Which of the following best summarizes how, in general, a nondihydropyridine CCB differs from nifedipine?
a. Causes a much higher incidence of reflex tachycardia
b. Causes significant dose-dependent slowing of AV nodal conduction velocity
c. Causes significant venodilation, leading to profound orthostatic hypotension
d. Has significant and direct positive inotropic effects
e. Is best used in conjunction with a β-blocker or digoxin
212. A patient has received excessive doses of nitroprusside, and toxic manifestations are developing in response to a metabolite. Which of the following drugs or drug groups would be most effective in limiting and ultimately counteracting the effects of the toxic product?
a. Aminocaproic acid
b. Hydroxycobalamin
c. Protamine sulfate
d. Thrombin
e. Vitamin K
213. A patient with Stage 2 essential hypertension is treated with usually effective doses of an ACE inhibitor. After a suitable period of time, blood pressure has not been lowered satisfactorily. The patient has been compliant with drug therapy and other recommendations (eg, weight reduction, exercise). A thiazide is added to the ACE inhibitor regimen. What is the most likely and earliest (and probably transient) untoward outcome of this drug add-on, for which you should monitor closely?
a. Fall of blood pressure sufficient to cause syncope
b. Hypokalemia due to synergistic effects of the ACE inhibitor and the thiazide on renal potassium excretion
c. Onset of acute heart failure from depression of ventricular contractility
d. Paradoxical hypertensive crisis
e. Sudden prolongation of the P-R interval and increasing degrees of heart block
214. A 45-year-old man postmyocardial infarction (MI) is being treated with several drugs, including intravenous unfractionated heparin. Stool guaiac on admission was negative, but is now 4+, and he has had an episode of hematemesis. What would be the best drug to administer to counteract the effects of excessive heparin remaining in the circulation?
a. Aminocaproic acid
b. Dipyridamole
c. Factor IX
d. Protamine sulfate
e. Vitamin K
215. A 45-year-old man asks his physician for a prescription for sildenafil to improve his sexual performance. Because of risks from a serious drug interaction, this drug should not be prescribed, and the patient should be urged not to try to obtain it from other sources, if he is also taking which of the following drugs?
a. Angiotensin-converting enzyme inhibitor
b. β-Adrenergic blocker
c. Nitrovasodilator (eg, nitroglycerin)
d. Statin-type antihypercholesterolemic drug
e. Thiazide or loop diuretic
216. A physician is preparing to administer a drug for which there is a label warning: “do not administer this drug to patients with second-degree or greater heart block, or give with other drugs that may cause heart block.” Which finding would be specifically indicative of heart block, and second-degree heart block in particular?
a. Auscultation of the precordium reveals an irregular rhythm
b. Blood pressure is low
c. Heart rate is abnormally low (bradycardia), but there is normal sinus rhythm
d. ECG reveals ventricular ectopic beats
e. ECG shows an excessively prolonged PR interval, and some P waves are not followed by a normal QRS complex
f. ECG shows abnormally widened QRS complexes
217. A 52-year-old woman with essential hypertension, hypercholesterolemia, and chronic-stable angina develops severe constipation. It is attributed to one of her medications. What was the most likely cause?
a. Atorvastatin
b. Captopril
c. Labetalol
d. Nitroglycerin
e. Verapamil
218. We use standard invasive hemodynamic techniques to measure or calculate the effects of various drugs on such parameters as arterial pressure, total peripheral resistance, and central venous (right atrial) pressures. Our goal is to evaluate whether the drug primarily causes arteriolar or venular dilation, or affects both sides of the circulation. Which drug exerts vasodilator effects only in the arterial side of the circulation?
a. Hydralazine
b. Losartan
c. Nifedipine
d. Nitroglycerin
e. Prazosin
219. A 20-year-old collegiate varsity hockey player is referred to you by his coach. The young athlete has excessive bruising after a very physical match 2 days before. His knee had been bothering him, so he took two 325 mg aspirin tablets several hours before the contest. He got checked hard into the boards many times during the game, but denies any excessive or unusual trauma. As you ponder the situation you order several blood tests. Which test or finding do you most likely expect to be abnormal as a result of the prior aspirin use?
a. Activated partial thromboplastin time (APTT)
b. Bleeding time
c. INR (International Normalized Ratio)
d. Platelet count
e. Prothrombin time
220. A patient in the coronary care unit develops episodes of paroxysmal AV nodal reentrant tachycardia (PSVT). What drug would generally be considered first-line for promptly stopping the arrhythmia?
a. Adenosine
b. Digoxin
c. Edrophonium
d. Phenylephrine
e. Propranolol
221. A 60-year-old man, hospitalized for an acute myocardial infarction, is treated with warfarin (among other drugs). What is the main mechanism by which warfarin is causing the effects for which it is given?
a. Increase in the plasma level of Factor IX
b. Inhibition of thrombin and early coagulation steps
c. Inhibition of synthesis/activation of prothrombin and Factors VII, IX, and X
d. Inhibition of platelet aggregation
e. Activation of plasminogen
f. Binding of Ca2+ ion cofactor in some coagulation steps
222. A 42-year-old man with an acute MI is treated with alteplase, and electrocardiographic and hemodynamic status improve quickly. By what mechanism did the alteplase cause its beneficial effects?
a. Blocked platelet ADP receptors
b. Inhibited platelet thromboxane production
c. Inhibited synthesis of vitamin K-dependent coagulation factors
d. Prevented aggregation of adjacent platelets by blocking glycoprotein IIb/IIIa receptors
e. Promoted conversion of plasminogen to plasmin
223. A patient with atrial fibrillation is placed on long-term arrhythmia control with amiodarone. In addition to “standard” monitoring, what should be assessed periodically in order to detect adverse effects that are rather unique to this drug among virtually all the antiarrhythmics?
a. Blood glucose, triglyceride, cholesterol, and sodium concentrations
b. Hearing thresholds (audiometry) and plasma albumin concentration
c. Prothrombin time and antinuclear antibody (ANA) titers
d. Pulmonary function and thyroid hormone status
e. White cell counts and blood urate concentration
224. A 64-year-old woman has had several episodes of transient ischemic attacks (TIAs). Aspirin would be a preferred treatment for prevention of thrombosis, but she has a history of severe “aspirin sensitivity” manifest as intense bronchoconstriction and urticaria. What would you consider to be the best alternative to the aspirin?
a. Acetaminophen
b. Aminocaproic acid
c. Clopidogrel
d. Dipyridamole
e. Streptokinase
225. A patient who has excessively slow AV nodal conduction rates, that unfortunately haven’t been recognized, is started on a drug. As soon as blood levels climb toward the usual therapeutic range the patient goes into complete heart block. Which drug most likely provoked this further prolongation of the P-R interval, ultimately leading to the cessation of all AV nodal conduction?
a. Captopril
b. Losartan
c. Nifedipine
d. Nitroglycerin
e. Prazosin
f. Verapamil
226. A patient with heart failure, Stage 1 essential hypertension, and hyperlipidemia (elevated LDL cholesterol and abnormally low HDL-C) is taking furosemide, captopril, atenolol, and simvastatin (an HMG-CoA reductase inhibitor).
During a scheduled physical exam, about a month after starting all the above drugs, the patient reports a severe, hacking, and relentless cough. Other vital signs, and the overall physical assessment, are consistent with good control of both the heart failure and blood pressure and indicate no other underlying disease or abnormalities. Results of blood tests are not yet available.
Which of the following is the most likely cause of the cough?
a. An expected side effect of the captopril
b. An allergic reaction to the statin
c. Dyspnea due to captopril’s known and powerful bronchoconstrictor action
d. Excessive doses of the bumetanide, which led to hypovolemia
e. Hyperkalemia caused by an interaction between bumetanide and captopril
f. Pulmonary edema from the bumetanide
227. A patient with a history of hypertension, heart failure, and peripheral vascular disease has been on oral therapy, with drugs suitable for each, for about 3 months. He runs out of the medication and plans to have the prescriptions refilled in a week or so.
Within a day or two after stopping his medications he experiences an episode of severe tachycardia accompanied by tachyarrhythmias, and an abrupt rise of blood pressure to 240/140 mm Hg—well above pretreatment levels. He complains of chest pain, anxiety, and a pounding headache. Soon thereafter he suffers a hemorrhagic stroke.
Which drug or drug group that the man was taking and stopped taking suddenly most likely caused these responses?
a. ACE inhibitor
b. Clonidine
c. Digoxin
d. Furosemide
e. Nifedipine (a long-acting formulation)
f. Warfarin
228. Your class just completed a minireview of the physiology and pathophysiology of blood pressure control and finally dawned on a classmate: bradykinin, an endogenous vasodilator, contributes to keeping blood pressure low; and its metabolite, formed by an enzyme cleverly called brady-kininase, lacks vasodilator activity. A colleague says “wouldn’t it be great if we had a drug that could inhibit bradykinin’s metabolic inactivation? We could probably use it as an antihypertensive drug!” You reply that we already have a drug—several, in fact—that does that very thing. What drug or drug group would that be?
a. Atenolol or metoprolol
b. Captopril or other “prils”
c. Hydrochlorothiazide or similar diuretic-antihypertensives
d. Labetalol or carvedilol
e. Losartan or other “sartans”
229. A 70-year-old woman is treated with sublingual nitroglycerin for occasional bouts of effort-induced angina. What best describes part of the mechanisms by which nitroglycerin causes its desired antianginal effects?
a. Blocks α-adrenergic receptors
b. Forms cyanide, much like the metabolism of nitroprusside does
c. Increases local synthesis and release of adenosine
d. Raises intracellular cGMP levels
e. Selectively dilates/relaxes coronary arteries
230. Your patient has bipolar illness, hypercholesterolemia, chronic-stable angina, and Stage 1 essential hypertension. He has been taking lithium and an SSRI for the bipolar illness. Cardiovascular drugs include atorvastatin, diltiazem, sublingual nitroglycerin, captopril, and hydrochlorothiazide. What outcome, due to interactions involving these drugs, should you most likely expect?
a. Development of acute psychosis from an ACE inhibitor-antipsychotic interaction
b. Development of a hypomanic state from antagonism of lithium’s action by the nitroglycerin
c. Lithium toxicity because of hyponatremia caused by the hydrochlorothiazide
d. Loss of cholesterol control from antagonism of the HMG Co-A reductase inhibitor by the antipsychotic
e. Worsening of angina because the antipsychotic counteracts the effects of the calcium channel blocker
f. Worsening of angina because the lithium antagonizes the effects of the nitroglycerin
231. A first-year house officer notices that a patient is experiencing significant and rapidly rising blood pressure (currently 180/120 mm Hg). One of the medications the patient had been taking is immediate-acting nifedipine oral capsules. There is a dose of this nifedipine formulation at the bedside, so the physician pricks the capsule open and squirts the contents into the patient’s mouth. This technique avoids “first-pass” metabolism of the drug and causes rapid absorption and all the effects associated with this calcium channel blocker. What is the most likely outcome of giving nifedipine as described here?
a. AV nodal block
b. Further rise of heart rate, worsening of the ventricular arrhythmia
c. Hypotension and bradycardia
d. Normalization of blood pressure and heart rate
e. Return of blood pressure toward normal, no significant effect on heart rate or the ECG
232. A 55-year-old patient with multiple cardiovascular diseases is being treated with digoxin, furosemide, triamterene, atorvastatin, and nitroglycerin—all prescribed by the family physician he’s had for decades. The patient now experiences nausea, vomiting, and anorexia, and describes a “yellowish-greenish tint” to white objects and bright lights. These signs and symptoms are most characteristic of toxicity due to which drug?
a. Atorvastatin
b. Digoxin
c. Furosemide
d. Nitroglycerin
e. Triamterene
233. A 28-year-old female patient has Stage 1 essential hypertension (resting BP 144/98), tachycardia, and occasional palpitations (ventricular ectopic beats). Normally we might consider prescribing a β-blocker to control the blood pressure and cardiac responses, but our patient also has asthma, and she is trying to get pregnant. Which drug would be the best alternative to the β-blocker in terms of likely efficacy on pressure and heart rate, and in terms of relative safety?
a. Diltiazem
b. Enalapril
c. Furosemide
d. Phentolamine
e. Prazosin
234. A patient presents with hypertension. The underlying cause—a pheochromocytoma—is not looked-for or detected in the initial work-up. An oral antihypertensive drug is prescribed. We soon find that the patient’s blood pressure has risen to levels above pretreatment levels—so much so that we are worried about imminently dangerous effects from the drug-induced worsening of hypertension—in response to a drug. Concomitant with the drug-induced rise of blood pressure the patient develops signs and symptoms of heart failure. Which drug was most likely administered?
a. Captopril
b. Hydrochlorothiazide
c. Labetalol
d. Losartan
e. Propranolol
f. Verapamil
235. A patient on long-term warfarin therapy has an INR that is excessive (4.5; normal, not anticoagulated is 1.0; the target for this patient was 2.5). She reports episodes of epistaxis over the last 2 days and now there is a great risk of serious bleeding episodes. In addition to stopping warfarin administration for a day or more, which drug would you want to administer to counteract warfarin’s excessive effects that led to spontaneous bleeding?
a. Aminocaproic acid
b. Epoetin alfa
c. Ferrous sulfate
d. Phytonadione (vitamin K)
e. Protamine sulfate
236. A patient with hypertension and heart failure has been treated for 2 years with carvedilol and lisinopril. He has just had hip replacement surgery, but because he is not ambulating he is started on unfractionated heparin, postoperatively, for prophylaxis of deep venous thrombosis. Oral antacids and esomeprazole (gastric parietal cell proton pump inhibitor) have been added for prophylaxis of acute stress ulcers. Five days postop, he experiences sudden onset dyspnea and electrocardiographic and other indications of an acute MI. The patient’s platelet counts are dangerously low. What is the most likely underlying problem?
a. Accidental substitution of low-molecular-weight heparins (LMWH) for unfractionated heparin
b. Accidental/inadvertent aspirin administration
c. Hemolytic anemia from a carvedilol-ACE inhibitor interaction
d. Heparin-induced thrombocytopenia
e. Reduced heparin effects by increased metabolic clearance (caused by ranitidine)
237. A patient with angina pectoris is started on a nitroglycerin transdermal delivery system (“skin patch”) for prophylaxis of his angina. He wears the patch 24 hours a day, 7 days a week, except for the few minutes when he showers each day. What is the main concern with “around-the-clock” administration of this or other long-acting formulations of nitrovasodilators?
a. Cyanide poisoning
b. Development of tolerance to their vasodilator actions
c. Gradual development of reflex bradycardia in response to successive doses
d. Onset of delayed, characteristic adverse responses including thrombosis and thrombocytopenia
e. Paradoxical vasoconstriction leading to hypertension
238. For many hypertensive patients we can prescribe either lisinopril (or an alternative in the same class) or losartan. What statement correctly summarizes how losartan differs from lisinopril other lisinopril-like drugs?
a. Lisinopril competitively blocks catecholamine-mediated vasoconstriction, losartan does not
b. Lisinopril effectively inhibits synthesis of angiotensin II, losartan does not
c. Losartan causes a higher incidence of bronchospasm and hyperuricemia
d. Losartan is preferred for managing hypertension during pregnancy, whereas captopril is contraindicated
e. Losartan is suitable for administration to patients with heart failure, whereas captopril and related drugs should be avoided.
239. A 46-year-old man has Stage 1 essential hypertension (resting BP 150/98), primary hypercholesterolemia, and modestly elevated fasting glucose levels (130 mg/dL) measured on several occasions. His cholesterol levels (total, HDL, LDL) have not been acceptably modified by dietary changes and daily use of a “statin.” The physician adds ezetimibe to the regimen. Which statement summarizes ezetimibe’s actions, or what would be expected in response to its use?
a. Exerts profound cardiac negative inotropic effects that poses a risk of heart block
b. Frequently causes orthostatic hypotension that in turn triggers reflex cardiac stimulation
c. More likely than other drugs to increase the risk of severe statin-induced myopathy
d. Reduces intestinal cholesterol uptake, has no direct hepatic effect to inhibit cholesterol synthesis
e. Significantly increases risk of atherosclerotic plaque rupture
240. A 58-year-old man presents in the emergency department with his first episode of acute coronary syndrome (ACS) and all evidence points to a myocardial infarction. Angioplasty and stenting are not possible because the cardiac cath lab is busy with other higher-priority patients, so administration of a thrombolytic drug is the only option. What is the most important determinant, overall, of the success of thrombolytic therapy in terms of salvaging viable cardiac muscle (or other ischemic tissues)?
a. Choosing a “human” (cloned) plasminogen activator (eg, t-PA), rather than one that is bacterial-derived (eg, streptokinase)
b. Infarct location (ie, anterior wall of left ventricular vs another site/wall)
c. Presence of collateral blood vessels to the infarct-related coronary artery
d. Systolic blood pressure at the time the MI is diagnosed
e. Time from onset of infarction to administration of the thrombolytic agent
241. A patient with an acute coronary syndrome is given a variety of cardiovascular drugs as he is being readied for transport to the “cath lab” for possible placement of a stent. One of the meds is abciximab. What best describes the mechanism of action of this drug?
a. Blocks thrombin receptors selectively
b. Blocks ADP receptors
c. Blocks glycoprotein IIb/IIIa receptors
d. Inhibits cyclooxygenase
e. Inhibits prostacyclin production
242. A patient presents in the emergency department with acute hypotension that requires treatment. Hypovolemia is ruled-out as a cause or contributor, and information gathered from the patient and family indicates that the cause is overdose of an antihypertensive drug.
One approach to treatment is to administer a pharmacologic (ordinarily effective) dose of phenylephrine, an α-adrenergic agonist. You do just that, and blood pressure fails to rise at all—and a second dose doesn’t work either. On which antihypertensive drug did the patient most likely overdose?
a. Captopril or another ACE inhibitor
b. Hydralazine
c. Prazosin
d. Thiazide diuretic (eg, hydrochlorothiazide)
e. Verapamil
243. An elderly man who has just been referred to your practice has been taking a drug for symptomatic relief of benign prostatic hypertrophy. In addition to its effects on smooth muscles of the prostate and urethra, this drug can lower blood pressure in such a way that it reflexly triggers tachycardia, positive inotropy, and increased AV nodal conduction. The drug neither dilates nor constricts the bronchi. It causes the pupils of the eyes to constrict and interferes with mydriasis in dim light. Initial oral dosages of this drug have been associated with a high incidence of syncope.
Which prototype is most similar to this unnamed drug in terms of the pharmacologic profile?
a. Captopril
b. Hydrochlorothiazide (prototype thiazide diuretic)
c. Labetalol
d. Nifedipine
e. Prazosin
f. Propranolol
g. Verapamil
244. You are contemplating starting ACE inhibitor therapy for a patient with essential hypertension. Which one of the following patient-related condition(s) contraindicates use of any ACE inhibitor and so should be ruled out before you prescribe this drug?
a. Asthma
b. Heart failure
c. Hyperlipidemia, coronary artery disease
d. Hypokalemia
e. Is a woman who is pregnant or may become pregnant
245. A patient develops sinus bradycardia. Heart rate is dangerously low, and an effective and safe drug needs to be given right away. Which drug would be the best choice for normalizing heart rate without initiating any other arrhythmias?
a. Atropine
b. Amiodarone
c. Edrophonium
d. Lidocaine
e. Phentolamine
246. A patient presents in the emergency department (ED) with severe angina pectoris, and acute myocardial ischemia is confirmed by electrocardiographic and other clinical indicators. Unknown to the ED team is the fact that the ischemia is due to coronary vasospasm, not to coronary occlusion with thrombi. Given this etiology, which drug, administered in usually effective doses, may actually make the vasospasm, and the resulting ischemia, worse?
a. Alteplase (t-PA)
b. Aspirin
c. Captopril
d. Nitroglycerin
e. Propranolol
f. Verapamil
247. Many clinical studies have investigated the benefits of daily aspirin use in the primary prevention of coronary heart disease and sudden death in adults. The results have been somewhat inconsistent, in part because different dosages were studied, and there were important differences in the populations that were studied. Nonetheless, many (if not most) of the studies have revealed that for some patients aspirin increased the incidence of a particularly unwanted adverse response, even when dosages were kept within the range typically recommended for cardioprotection (eg, 81-162 mg/day). What is the most likely adverse response associated with aspirin prophylaxis, particularly in patients who have a low risk of an acute coronary syndrome or cardiovascular disease in general?
a. Centrolobular hepatic necrosis
b. Hemorrhagic stroke
c. Nephropathy
d. Tachycardia and hypotension leading to acute myocardial ischemia
e. Vasospastic angina
248. A patient with essential hypertension has been treated with a fixed-dose combination product that contains hydrochlorothiazide and triamterene. Blood pressure and electrolyte profiles have been kept within acceptable limits for the last 18 months. Now, however, blood pressure has risen to the point where the physician wants to add another antihypertensive drug. The drug is started; after several weeks blood pressure falls into an acceptable range, but the patient has become hyperkalemic. What drug was added and was most likely responsible for the desired blood pressure fall and the unwanted rise of potassium levels.
a. Diltiazem
b. Prazosin
c. Propranolol
d. Ramipril
e. Verapamil
249. A patient has a supraventricular tachycardia. We inject a drug and heart rate falls to a normal (or at least more acceptable) level. Although this drug caused the desired response, it did so without any direct effect in or on the heart. Which drug was most likely used?
a. Edrophonium
b. Esmolol
c. Phenylephrine
d. Propranolol
e. Verapamil
250. A 69-year-old man presents with NYHA Stage II (“mild”) heart failure. His symptoms failed to improve adequately in response to captopril and carvedilol so the physician stops the carvedilol and adds usual therapeutic doses of digoxin and furosemide. At a follow-up exam 3 months later we find good symptomatic relief of the heart failure. Blood electrolytes and all other lab tests are within normal limits. At this time, which electrocardiographic change would you expect to see in response to the digoxin, compared with a baseline (pretreatment) ECG?
a. P waves widened, amplitude increased
b. PR intervals prolonged
c. QRS complexes widened
d. RR intervals shortened
e. ST segments elevated
251. A patient presents with a blood pressure of 220/120 and a heart rate of 90 beats per minute despite usually effective antihypertensive drug therapy. Further work-up indicates the patient has a rare cause of these and other signs and symptoms: pheochromocytoma. You realize that β-adrenergic blockers are useful as antihypertensive drugs, and for helping to normalize heart rate in patients with supraventricular tachycardia. As a result of the diagnosis, and your knowledge, you administer a usually effective dose of propranolol. What is the most likely outcome of doing this?
a. Blood pressure falls promptly, followed by reflex tachycardia
b. Epinephrine release from the tumor is suppressed, hemodynamics normalize
c. Heart rate and cardiac function rise quickly because the β-blocker has triggered additional epinephrine release from the tumor.
d. Left ventricular afterload is decreased, cardiac output rises via increases of both left ventricular stroke volume and heart rate
e. Total peripheral resistance rises, cardiac output falls, the patient goes into cardiogenic shock
252. A 59-year-old man presents in the emergency department with crushing chest discomfort. An ECG indicates a small transmural left ventricular infarction, and prompt cardiac catheterization and assessment of prior lab results indicate significant hypercholesterolemia. The patient is given all the drugs listed below, for both immediate management of the ischemia and its symptoms and for long-term prevention of a subsequent, and potentially fatal, MI. Which drug would provide immediate relief of the consequences of myocardial ischemia, but has no long-term effects to reduce the risk of sudden death or ventricular dysfunction from another MI?
a. Aspirin
b. Atorvastatin
c. Captopril
d. Nitroglycerin
e. Propranolol
253. A 65-year-old woman is transferred to the thoracic surgery ICU after cardiac surgery. She has diffuse rales bilaterally, a pulse of 90/min, an elevated central venous pressure, and a blood pressure of 160/98 mm Hg. The surgery resident wants to inject an otherwise-correct dose of an IV drug to control heart rate and blood pressure, but grabs a syringe that contains another drug. The patient’s heart rate increases to 150/min and her blood pressure rises to 180/106. Which drug did this patient most likely receive in error?
a. Dobutamine
b. Esmolol
c. Neostigmine
d. Propranolol
e. Verapamil
254. A patient with chronic-stable (“effort-induced”) angina begins taking metoprolol, and once blood levels approach the therapeutic range the frequency and severity of angina attacks, and the need for sublingual nitroglycerin, were reduced. Which of the following states the direct pharmacologic action by which the β-blocker provided the desired effects?
a. Decreased myocardial oxygen demand
b. Dilated the coronary vasculature
c. Directly inhibited angiotensin II synthesis
d. Reduced total peripheral resistance
e. Slowed AV nodal conduction velocity
255. A 50-year-old man is aware of the benefits of aspirin in terms of reducing the risk of death from an acute myocardial infarction, mainly because he has seen many of the ads and internet posts about this. He notices that the usual recommended dose of aspirin for cardioprotection is 81 mg/day, but reasons that the bigger the dose, the bigger and better the protective effect. He has taken “at least” 1,000 mg of aspirin (3 “regular strength” aspirin tablets) twice a day for the last 6 months. While he is fortunate in terms of having no apparent gastrointestinal adverse effects that are associated with long-term, high-dose aspirin use, he suffers an MI. Autopsy results show considerable platelet occlusion of several coronary vessels. What explains the most likely mechanism by which high dose aspirin use these adverse events?
a. Inhibited thromboxane A2 synthesis in platelets
b. Favored adhesion of platelets to the vascular (coronary) endothelium
c. Ruptured atherosclerotic plaque in the coronaries, exposing platelets to collagen
d. Suppressed hepatic synthesis of vitamin K-dependent clotting factors
e. Triggered excessive activation of platelets by ADP
256. In clinic you meet a 55-year-old man who is described by the attending as having “metabolic syndrome,” including high LDL and low HDL cholesterol levels, essential hypertension, type 2 diabetes mellitus, and anginal attacks upon stress about once every 2 months. He currently has asymptomatic hyperuricemia, but has a gout attack about once a year. The patient is obese (92 kg), 6 feet tall, and has a body mass index (kg/square meter of body surface area) of 40 (normal or desirable no more than 24.9 kg/m2). He has a 20 year history of smoking a half pack of cigarettes a day, and both parents died in their late 50s—the father from an acute MI, the mother from hemorrhagic stroke. The gentleman is taking medications deemed appropriate for each of the conditions noted above. One is colesevelam. What is the probably reason why the colesevelam was given?
a. Counteracts hypokalemia caused by a thiazide diuretic
b. Lowers LDL-cholesterol levels
c. Lowers plasma urate levels, prophylaxis of gout
d. Prevents myocardial ischemia, angina, by reducing myocardial oxygen demand
e. Provides antihypertensive and natriuretic effects
257. A man has an aneurysm in the aortic root, a consequence of Marfan syndrome. He experiences a hypertensive crisis that requires prompt blood pressure control. Nitroprusside will be infused for its immediate antihypertensive effects. What drug would we administer along with the nitroprusside to minimize the risk of aneurysm rupture due to increases of left ventricular dP/dt (ΔP/Δt; change in pressure/change in time) as blood pressure falls?
a. Atropine
b. Diazoxide
c. Furosemide
d. Phentolamine
e. Propranolol
258. Nicotinic acid (niacin), in the relatively large doses that are used to treat certain and common hyperlipidemias, often causes a cutaneous flush and pruritus. These can be accompanied by widely distributed and sharp “pins and needles” or burning sensation on the skin. The response can be attenuated by several means, one of which involves pretreatment with aspirin. What mechanism or action most likely contributes to the vasodilatory response and the flushing?
a. Activation of α-adrenergic receptors on vascular smooth muscle
b. Calcium channel blockade in vascular smooth muscle
c. Local production of prostaglandins
d. Release of angiotensin II
e. Release of histamine
259. The figure below shows typical cardiovascular responses to the slow IV injection of four adrenergic drugs into a normal, resting subject. Assume the doses of each are sufficient to cause the effects seen here, but not so high that toxic effects occur. No other drugs are present, and sufficient time has been allowed to enable complete dissipation of the effects of any prior drugs. The dashed line between the systolic and diastolic pressure traces approximates mean arterial pressure.
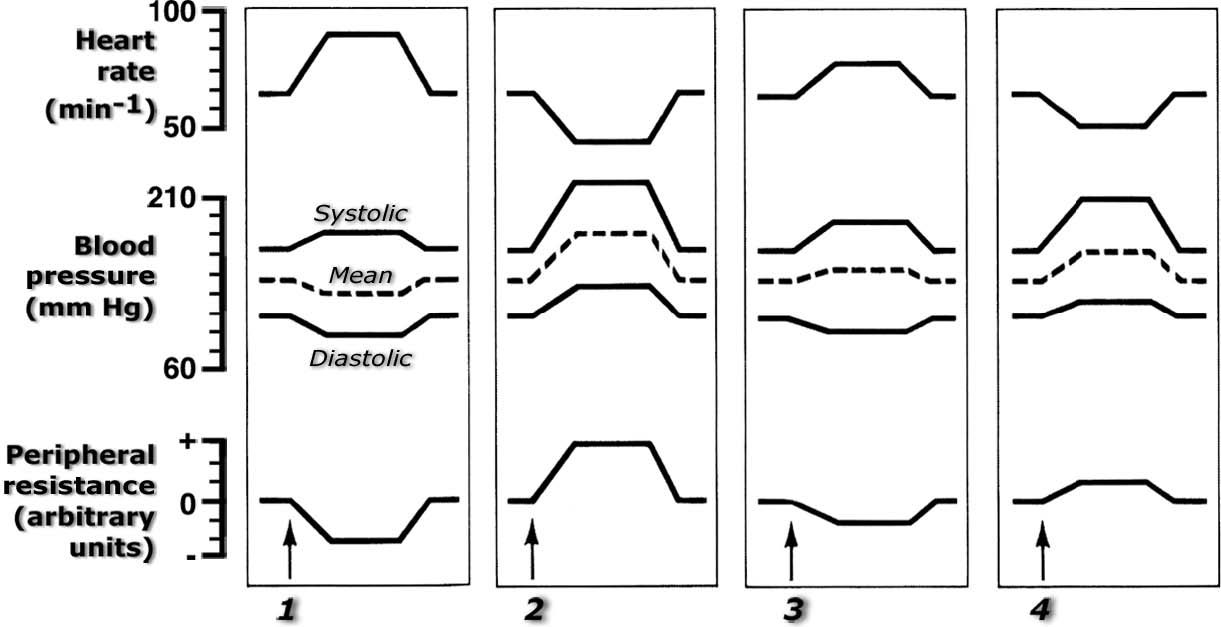
Abbreviations used, and answer choices, are
EPI, epinephrine
ISO, isoproterenol
NE, norepinephrine
PHE, phenylephrine
PHN, phentolamine
PRO, propranolol
Select the letter that indicates the drugs that are ordered in the sequence shown (1, 2, 3, 4).
a. EPI, NE, PHE, ISO
b. ISO, EPI, NE, PHE
c. ISO, PHE, EPI, NE
d. NE, ISO, PHE, EPI
e. PHE, EPI, NE, PRO
f. PHE, ISO, NE, EPI
g. PRO, PHN, PHE, ISO
Look at the ECG below and answer the following three questions.
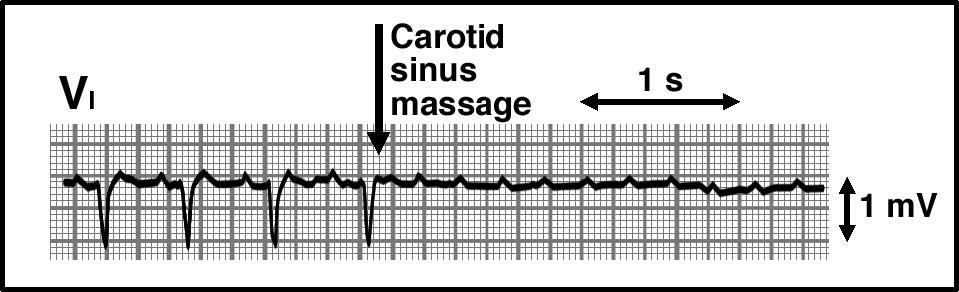
Here you see a continuous (uninterrupted) tracing of lead V1, before and after carotid sinus massage (at arrow).
260. What is the mechanism by which carotid massage exerted its effect?
a. Activated what is tantamount to the baroreceptor reflex, increasing vagal tone and acetylcholine release considerably
b. Caused catecholamine release
c. Induced atrial fibrillation (atrial rate >> 300/min)
d. Occluded venous return to the heart, thereby interfering with filling and contraction of all heart chambers “downstream” of the right atrium
261. Based on the outcome of carotid sinus massage, what can you say about the origin of the aberrant electrical activity that leads to the tachycardia you see before the massage?
a. Bundle of Kent (ie, anomalous or accessory pathway for AV conduction) with retrograde and antegrade conduction
b. Left bundle branch
c. Multiple ectopic ventricular foci
d. Supraventricular
262. What drug, given as an intravenous bolus, might be used as an alternative to carotid massage, causing essentially the same outcome and, therefore, the same interpretation of the origin of the ventricular tachycardia?
a. Adenosine
b. Atropine
c. Epinephrine
d. Isoproterenol
e. Lidocaine
263. A 23-year-old nonpregnant woman has been using a preparation of oral ergotamine to manage her frequent migraine headaches. She consumes an excessive dose of the drug while trying to abort a particularly severe and refractory attack. What adverse cardiac or cardiovascular consequences are most likely to occur as a result of the ergot overdose?
a. Myocardial and peripheral (eg, limb) ischemia due to intense vasoconstriction
b. Renal failure secondary to rhabdomyolysis
c. Spontaneous bleeding due to direct inhibition of platelet activation/aggregation
d. Syncope secondary to acute hypotension
e. Tachycardia, tachyarrhythmias from β-1 adrenergic receptor activation
Cardiovascular Pharmacology
Answers
178. The answer is a. (Brunton, pp 730-736; Katzung, pp 184-185, 219-220, 295-300.) ACE inhibitors are often selected first for hypertensive patients who also have diabetes mellitus (type 1 or type 2)—provided their renal function is satisfactory (specifically, no severe bilateral renal arterial stenosis, or no inadequate blood flow to one kidney if the other was removed; no albuminuria). Recall that angiotensin II constricts the efferent arterioles in the kidneys, which increases glomerular filtration. So, by reducing angiotensin II synthesis an ACE inhibitor will block this effect and reduce filtration even further. Angiotensin receptor blockers (ARBs, eg, losartan) will do the same but by blocking angiotensin receptors rather than by inhibiting angiotensin synthesis.
ACE inhibitors (and ARBs) do not cause any problems with glycemic control or the responses to antidiabetic drugs, and they seem to exert some protective effect that slows or delays diabetes-related nephropathy. Note that either class of angiotensin modifier is contraindicated for patients with severe bilateral renal arterial stenosis, including that which might occur with long-standing and severe diabetes mellitus. In such patients, maintenance on some reasonable degree of renal perfusion and renal function overall depends on the renovascular/postglomerular vasoconstrictor effects of angiotensin. Block angiotensin synthesis (ACE inhibitors) or angiotensin’s ability to activate its receptors (ARBs), and acute renal failure may ensue.
β-Adrenergic blockers would not be a good choice if hypertension is accompanied by diabetes mellitus, particularly when insulin is used for glycemic control. Should the diabetic patient experience an episode of hypoglycemia, a β-blocker may delay recovery of blood glucose levels, mask tachycardia that is one symptom of hypoglycemia development, and interact with some antidiabetic drugs (even those used for type 2; to cause excessive blood glucose-lowering). (If the diabetes is well controlled, and if the patient has other disorders for which benefits of β-blockade may outweigh potential problems, such as mild-moderate heart failure or recent myocardial infarction, then a β-blocker may be considered.)
Thiazides can elevate blood glucose levels and antagonize the desired effects of antidiabetic drugs (probably by reducing the responsiveness of skeletal muscle cells and adipocytes to insulin). Nonetheless, they might not be a first-choice in the setting of diabetes—at least not for the patient described in the scenario. (Some clinicians may disagree, and consider a thiazide or thiazide-like diuretic (eg, metolazone) a first-choice drug for essential hypertension in most patients.)
Verapamil, diltiazem, and nifedipine seem not to complicate blood glucose regulation or interact with antidiabetic drug therapy. Nonetheless, they lack other benefits offered by ACE inhibitors (especially the renal-protective effects) in the setting of diabetes and so would not be a first choice or of special value.
179. The answer is d. (Brunton, pp 774t, 780-781; Katzung, pp 155-162, 167, 179.) Labetalol is the best choice, assuming no contraindications to β-blockade, such as asthma or second degree or complete heart block. Given its combination of both α-and β-adrenergic (β1 and β2) blocking effect, it offers the best approach for managing the hypertension, the tachycardia, the resulting oxygen supply-demand imbalance that leads to both chest discomfort and the ischemic ST-changes, and the ventricular ectopy (which is probably a reflection of excessive catecholamine stimulation of β1-receptors). If the patient is having an acute myocardial infarction, starting β-blocker therapy early is also decidedly beneficial short term and for the long run. (Most any other β-blocker might be a suitable alternative, but labetalol has the combined α/β-blocking actions that are likely to be of greatest benefit. Carvedilol has the same profile, but it is given orally and in this setting that would not be ideal because of slow onset of action.)
Aspirin will do no harm in this situation, but it will also do no good acutely unless there is ongoing platelet aggregation and coronary occlusion. Even if there were, the aspirin would do little to control heart rate, blood pressure, or the ECG changes.
Nothing in the scenario suggests that this patient is volume-overloaded or suffering acute pulmonary edema. Therefore, administering the furosemide (c) in such a situation is not appropriate. Moreover, giving it is likely to cause prompt reductions of blood volume and, along with it, of blood pressure. The latter effect is likely to lead to further—and unwanted—reflex sympathetic activation that would make matters worse.
Lidocaine (e) might be suitable for the ventricular ectopy. However, I have identified several other important signs and symptoms that would not be relieved by this antiarrhythmic drug. As noted above, the profile of labetalol offers the greatest likelihood of managing multiple problems with one drug.
Increasing the dose of nitroglycerin (f; and especially giving it as a bolus) is likely to drop blood pressure acutely, triggering reflex (baroreceptor) stimulation of the heart. The usual “anti-ischemic” effects of the drug would be counteracted by such “pro-ischemic” changes as further rises of heart rate and a probable worsening of the premature ventricular beats.
Prazosin (g) would lower blood pressure nicely. However, once again we have to worry about excessive pressure lowering, triggering the baroreceptor reflex, and worsening many of the already worrisome findings (eg, heart rate, PVCs).
180. The answer is d. (Brunton, pp 313t, 326, 767, 774t, 831; Katzung, pp 159t, 161, 167, 189.) Timolol is a nonselective β-adrenergic blocker. Oral dosage forms are approved for managing essential hypertension (and angina). A topical ophthalmic dosage form is indicated for managing some cases of chronic open-angle glaucoma. β-Blockers are not only likely to control blood pressure, and help lower intraocular pressure, but also to reduce catecholamine-induced cardiac stimulation (the tachycardia).
Verapamil or diltiazem (b, e), both calcium channel blockers (CCBs) of the nondihydropyridine class, will not only lower blood pressure but also tend to modulate the tachycardia (through direct and β-receptor-independent processes). However, they don’t lower intraocular pressure. All other things being equal, then, the β-blocker would still be a better choice for the hypertensive, tachycardic, glaucomatous patient. Thus, we get three potential benefits from β-blockade and only two with verapamil or diltiazem.
ACE inhibitors, such as captopril (a) and thiazide diuretics (c), have no actions that would make them particularly suitable for hypertensive patients who are tachycardic, have glaucoma, or both.
181. The answer is c. (Brunton, pp 330, 780-781; Katzung, pp 159t, 161, 179-180.) Even if you know nothing specifically about nebivolol you should be able to conclude that increased formation or activity of nitric oxide (a vasodilator, among other things) is the only reasonable answer. A drug that competitively blocks α1 receptors (a; for example phentolamine or prazosin) would, indeed, lower blood pressure (BP). However, the question stated that nebivolol had no effect on the vasoconstrictor effects of either phenylephrine (direct α-agonist) or amphetamines (indirect-acting sympathomimetics; release neuronal norepinephrine). (Recall that labetalol and carvedilol are the β-blockers that also block α1 adrenergic receptors.) Competitive blockade of α2 receptors (b) cannot be correct. Blocking the presynaptic α-receptors would inhibit released norepinephrine’s usual ability to “feedback inhibit” further NE release upon an action potential. Had nebivolol blocked the α2 receptors, then, the most likely response would be a rise, not a fall, of peripheral resistance and BP. Thromboxane A2 (d) is a vasoconstrictor (among other things). Increasing its synthesis would tend to raise BP/peripheral resistance also. COMT (e) inhibits the metabolic inactivation of catecholamines in the GI tract, liver, and elsewhere. In theory, COMT inhibitors also would raise BP caused by its substrates. Finally, β-blockers are not catecholamines, and so are not substrates for COMT.
182. The answer is a. (Brunton, pp 86t, 1271-1272; Katzung, 743, 762-763, 1033t.) β1 activation, whether in contractile, nodal, or conducting tissues of the heart, leads to and requires increased production of cyclic AMP. Those responses will be inhibited in the presence of excessive doses of a β-blocker. Glucagon, a parenteral drug normally used for managing severe and acute hypoglycemia, can directly increase c-AMP levels in the heart, even in the presence of β-blockade. It has the best chance of helping to restore (or at least improve) cardiac function—even in the presence of profound β-blockade.
Phenylephrine (b), the prototype α-agonist, would either not affect the already high TPR, or might increase it further. Giving it in this case would not only be irrational, but also dangerous. Phentolamine (c), the prototype α-antagonist, would lower TPR, but it is still unlikely that the heart can generate sufficient CO to perfuse the tissues adequately. Ephedrine (d), a drug that works in part by releasing norepinephrine, would be of little if any benefit. If it were given CO would still be depressed owing to the β-blockade, and α-receptor activation would cause phenylephrine-like effects on the peripheral vasculature. As noted above, those peripheral vasoconstrictor effects are precisely what we don’t need or want to cause. Epinephrine (e) would also cause not-needed/unwanted peripheral vasoconstriction; it is not likely to do better at activating cardiac β-receptors, and improving CO, than isoproterenol already has.
183. The answer is e. (Brunton, pp 844, 1405-1407; Katzung, pp 238, 921-922.) Many of the signs and symptoms of salicylism are similar to those caused by high blood levels of quinidine (antiarrhythmic, no longer a first-line antiarrhythmic) or quinine (mainly used as an antimalarial). Quini-dine, quinine, and related drugs are called cinchona alkaloids, and the low grade toxicity syndrome caused by these drugs is called cinchonism. (These drugs were originally obtained from a plant known, generically, as Cinchona.) Aspirin and the cinchona alkaloids are chemically similar in some important chemical and pharmacologic ways. The common signs and symptoms include light-headedness, tinnitus, and visual disturbances such as diplopia.
184. The answer is e. (Brunton, pp 761, 765-767, 829-831; Katzung, pp 201-205, 245.) Verapamil, a nondihydropyridine calcium channel blocker (CCB), depresses both the SA node and the AV node and would be effective for prophylaxis of paroxysmal atrial or supraventricular tachycardia. Nifedipine (c), the prototypic dihydropyridine CCB, has little effect on SVT because it and the other dihydropyridines lack cardiac depressant effects. Moreover, if we chose a fast-/immediate-acting dosage form of nifedipine, we would probably trigger substantial reflex cardiac stimulation. The increased sympathetic tone to the heart could worsen the PSVT. Nitroglycerin (d) is mainly a venodilator, but it can cause falls of arterial blood pressure sufficient to trigger reflex cardiac activation that would exacerbate the tachycardia. Adenosine (a) may be useful in diagnosing whether ventricular tachycardia is of supraventricular or ventricular origin, because it effectively slows AV nodal conduction. It is also used as acute therapy for PSVT. However, it is a fast- and short-acting parenteral drug, which renders it unsuitable for the condition I stated: outpatient prophylaxis. Lidocaine (b) is mainly used for ventricular tachyarrhythmias, not those of supraventricular origin. Both adenosine and lidocaine are parenteral drugs with short half-lives; neither property makes them suitable for outpatient prophylactic use.
185. The answer is d. (Brunton, pp 206t, 212, 305-307, 309; Katzung, pp 151-152, 154, 167, 180, 189.) Prazosin causes competitive α-blockade. This is in stark contrast with phenoxybenzamine, which causes insurmountable (noncompetitive) and long-lasting blockade of α-receptors by alkylating them. Phenoxybenzamine is classified as a haloalkylamine. You probably don’t need to remember that, but perhaps it will help you realize that the drug alkylates the α-adrenergic receptors. This covalent interaction, in comparison with typical weak ionic interactions between most drugs and their receptors, accounts for phenoxybenzamine’s long-lasting (not shorter-acting; a) and noncompetitive blockade. As a result of altering receptor confirmation, not merely occupying them as prazosin and most other α-blockers do, phenoxybenzamine renders largely ineffective α-agonists that ordinarily would be used to counteract effects of excessive receptor blockade. Thus, if the patient became hypotensive in response to phenoxybenzamine, raising pressure with typical and usually effective drugs may not succeed. Neither drug has intrinsic β-blocking activity (b), and so regardless of which of these drugs we use for the pheochromocytoma patient, a β-blocker will need to be used adjunctively to control (primarily) heart rate and contractility. Neither drug suppresses epinephrine release from the adrenal/suprarenal medulla (c); both tend to cause orthostatic hypotension by blocking peripheral α-mediated vasoconstriction that occurs upon standing up suddenly.
186. The answer is c. (Brunton, pp 859, 864-865; Katzung, pp 604-607.) This brief scenario describes heparin-induced thrombocytopenia (HIT). It is an immune-mediated thrombocytopenia that is accompanied by a paradoxical increase in thrombotic events (eg, in limbs, brain, lungs, and heart). It affects about 1% to 3% of patients receiving heparin for more than about 4 days in a row. The cause appears to involve formation of antibodies that develop to heparin-platelet complexes. This leads to substantial increases of platelet activation that, in turn, leads to thrombosis, vascular damage, and eventually significant declines in the number of functional circulating platelets (since platelets have been consumed in widespread thrombotic events). This phenomenon is not associated with any of the other drugs listed in the question.
Important note: Warfarin can also cause a paradoxical prothrombotic state, probably due to suppressed protein C activity. It is most likely to occur if loading doses are used, rather than starting with the anticipated maintenance dose. If you do some checking on the web or in medicine texts, you will probably find several algorithms for calculating warfarin loading doses. The safest advice is not to use any loading doses: always get the initial anticoagulation with a heparin, start warfarin simultaneously, then continue the heparin for at least 5 days and until the INR reaches the desired range. Using warfarin loading dose strategies does not hasten warfarin’s antithrombotic effect.
187. The answer is d. (Brunton, pp 801-804, 837-839; Katzung, pp 215-218, 220.) Digoxin inhibits the sarcolemmal Na+, K+-ATPase (“sodium pump”). This reduces the active (ATP-dependent) extrusion of intracellular Na+. The relative excess of intracellular Na+ competes with intracellular Ca2+ for sites on a sarcolemmal 2Na-Ca exchange diffusion carrier, such that less Ca2+ is extruded from the cells. The net results include a rise of free [Ca2+]i and greater actin-myosin interactions (ie, a positive inotropic effect that increases cardiac output through an increase of stroke volume).
188. The answer is a. (Brunton, pp 752, 767-768; Katzung, pp 205-207.) β-Blockers (represented in this question by atenolol) should not be administered (especially by a systemic route) to patients with vasospastic angina unless for a medical emergency that requires β-blockade as a life-saving measure. Recall the dual roles of adrenergic receptors in the coronary vasculature. Activation of β2-adrenergic receptors causes vasodilation. Activation of α-adrenergic receptors in the coronary (and other) vasculature favors vasoconstriction or—in the setting of variant angina—vasospasm. Normally these receptors are exposed to circulating epinephrine, which causes the opposing vasodilator (β2) and vasoconstrictor (α) effects. Norepinephrine is also activating α-adrenergic receptors. Block only the β2 (vasodilator) effects in the coronaries and the constrictor (and spasm-favoring) effects of the α receptors are left unopposed.
Diltiazem (b), a nondihydropyridine calcium channel blocker, would not only lower blood pressure but also suppress the tendency for coronary vasoconstriction or spasm by blocking vascular smooth muscle calcium influx. Hydrochlorothiazide (c) is the prototype thiazide diuretic, and metolazone (e) is thiazide-like in terms of most of its pharmacologic profiles. Neither is at all likely to cause or favor vasoconstriction in the coronary vessels or elsewhere. Losartan (d) is an angiotensin receptor blocker. It not only has good antihypertensive activity, but also is apt to suppress any coronary vasoconstrictor influences of circulating angiotensin.
189. The answer is c. (Brunton, pp 753-755, 760; Katzung, pp 195-201.) At usual therapeutic doses, nitroglycerin (NTG) acts primarily as a peripheral venodilator. At higher doses arterial dilation also occurs. The net effect is that, particularly with excessive doses, peripheral vasodilation occurs and blood pressure (BP) falls. Critical among these pressure changes is a fall of diastolic BP. I say critical because DBP is the main driving force for blood flow through the coronary vasculature. Up to a point autoregulation of coronary flow may help maintain cardiac perfusion, but once DBP reaches a certain lower level coronary blood flow is inadequate to meet the oxygen demands of the myocardium (ie, ischemia has developed). Add to the underperfusion of the heart by reduced DBP the fact that if BP falls by a sufficient amount, and sufficiently fast, the baroreceptor reflex is activated. That, in turn, increases heart rate and contractility—an increase of myocar-dial oxygen demand simultaneous with reduced oxygen supply due to the decreased coronary perfusion.
Nitroglycerin is not metabolized to cyanide (a). If you selected that possibility as your preferred answer you are confusing NTG with another nitrovasodilator that clearly does form cyanide: nitroprusside. NTG does not, cannot, induce coronary vasoconstriction or spasm directly (b). NTG is often used, with success and no extra risk, for patients with coronary vasospasm (d). For such individuals NTG is mainly used for acute management of an ongoing attack (as applies to virtually all angina patients who take the drug), with long-term suppression of vasospasm provided by a calcium channel blocker. Finally, for patients with angina that does not involve vasospasm, NTG is frequently prescribed with a β-adrenergic blocker (e). Here again, the NTG is used mainly for acute symptom control and the β-blocker for prophylaxis.
As an aside, if you indeed chose answer e, that the β-blocker’s “intrinsic vasodilator activity potentiated that of the NTG,” I think you should be prepared to cite a specific β-blocker with that property. The β-blocker named in the question, propranolol, has absolutely no intrinsic vasodilator activity. Indeed, of the long list of β-blockers on the market, only three have the ability to cause vasodilation. Labetalol and carvedilol have some α-blocking activity that causes vasodilation, but the effect is weak. Then there is nebivolol, which causes some vasodilation through its metabolism to nitric oxide.
Note: There is an interesting and somewhat controversial phenomenon ascribed to NTG and some other vasodilator drugs: coronary steal. In essence, vasodilation shunts or diverts (steals) blood flow from areas of low perfusion (eg, a stenotic coronary vessel) to areas of high perfusion. The postulated outcome is that poorly perfused vessels (and the tissues served by them) become even less well perfused, thereby causing or worsening distal tissue ischemia.
190. The answer is a. (Brunton, pp 688t, 774-775, 791; Katzung, pp 260-262, 270.) Thiazide diuretics tend to raise uric acid concentration in the blood, probably by inhibiting tubular secretion of urate. The elevations of urate may be of little concern for patients with no history of hyperuricemia or gout, but for those with such a history it can be a problem that is not associated with any of the other answer choices given. Thiazides can be administered to hyperuricemic/gouty patients, but that may require another drug (allopurinol or febuxostat, which are xanthine oxidase inhibitors) to counteract diureticinduced rises of urate levels. If one can avoid the problems by avoiding the thiazide, and the possible need for adding a second drug to counteract the hyperuricemia, why not do just that? That is one of many reasons why we might use another class of antihypertensive drugs (eg, angiotensin modifiers, calcium channel blockers, many more) that don’t have negative effects on urate levels.
None of the other answer choices have any appreciable desired or untoward effects on urate levels, renal handling of urate, or the incidence or severity of gout.
191. The answer is d. (Brunton, pp 849, 946, 949, 977-982; Katzung, pp 320, 612.) These in vitro responses, showing that ibuprofen (or another NSAID, naproxen) can significantly antagonize the antiplatelet effects of aspirin, are clinically relevant. Both ibuprofen and aspirin compete for the same binding sites on both COX-1 and COX-2. Aspirin’s interaction is covalent, and irreversible. Ibuprofen, however, binds to the same COX sites as aspirin. Its antiplatelet effects are comparatively weak and reversible. More importantly, the bound ibuprofen prevents the ability of aspirin to bind to same critical enzyme site. If aspirin therapy is stopped, COX-1 activity will increase by about 10% per day, and just 20% of baseline COX activity—that is, skipping aspirin for more than 2 to 3 days—may be enough to enable sufficient COX activity to be restored and the protective antiplatelet effects lost. Using ibuprofen for more than a few days in a row is likely to do the same. In short, the interaction should be avoided. And remember: ibuprofen (and naproxen) is available OTC, and unless you explicitly tell your patients to avoid it, your pharmacologic efforts to afford cardioprotection may be for naught. Such NSAIDs as diclofenac, and many others, do not interfere with aspirin’s antiplatelet effects.
Acetaminophen (a), in dosages of about 1 g/day or more, taken for more than 7 days in a row, may potentiate warfarin’s anticoagulant effects. However, it does not interfere with aspirin’s antiplatelet effects; and it has no anticoagulant or antithrombotic effects in its own right. Clopidogrel (b), a platelet ADP receptor blocker that is used as an antiplatelet drug in lieu of aspirin (for some patients), does not block aspirin’s antiplatelet effects, because the mechanisms of action of clopidogrel and aspirin are synergistic and different. Dabigatran (c) is an oral anticoagulant, sometimes used as an alternative to warfarin (e). It is classified as a direct-acting thrombin inhibitor. Neither dabigatran nor warfarin counteracts aspirin’s antiplatelet effects.
192. The answer is e. (Brunton, pp 752-753, 885, 892-898; Katzung, pp 625-627.) The findings are consistent with statin-induced myositis and myopathy, which seem to have progressed to rhabdomyolysis and renal failure—both potentially fatal. This syndrome (and hepatotoxicity) is the most serious adverse response to the statins. It is more prevalent in older patients, those with multiple illnesses, and especially those with renal or liver disease. Coadministration of most other lipid-lowering drugs (none of which are in the above list) increases the risk of rhabdomyolysis, hepatotoxicity, or both. As an aside, the risk of rhabdomyolysis (or lesser skeletal muscle changes) is not too much different between the currently available statins. However, it was (allegedly) such a problem with one relatively recent drug, cerivastatin, that the drug was pulled from the market.
Note: Never been taught specifically about rosuvastatin? Well, the drug name ends in “-statin” so you should automatically know quite a bit about it.
193. The answer is a. (Brunton, pp 178-179, 214t; Katzung, pp 98, 103, 331.) ACh causes vasodilation only when the vascular endothelium is intact functionally and anatomically. The normal response, initiated by ACh acting as an agonist on muscarinic receptors, involves endothelium-derived relaxing factor/nitric oxide (EDRF/NO) that is generated in and released from the endothelium (that is why it’s called “endothelium-derived”). The NO diffuses into the adjacent vascular smooth muscle cells and increases cGMP formation. That, in turn, causes extrusion of Ca2+ and relaxation of the muscle (vasodilation in the intact animal). When the endothelium is removed (actually quite easy to do experimentally) or damaged, ACh causes concentration-dependent increases of smooth muscle tension.
Isoproterenol (b) is a β1/β2 agonist. Added to an in vitro preparation such as the one used here, it would only cause smooth muscle relaxation (β2 effect) unless the β-receptors were blocked; it does not alter the contractile responses to added ACh. Blockade of muscarinic receptors (c) cannot be correct. If that were the case, particularly under control conditions, contractile response to ACh would be diminished or abolished, and contraction shown under the experimental conditions would not be changed from what is shown in the figure. Prazosin (d) cannot be correct. It is an α1 receptor blocker, and pretreating with it would not cause vasoconstriction with or without added ACh. Botulinum toxin (e) blocks ACh release from cholinergic nerves. Although there may be some postganglionic para-sympathetic (and sympathetic) nerve “endings” in the tissue sample, the responses shown above were caused by adding ACh directly to the tissue bath and so they are independent of any residual autonomic neural influences.
194. The answer is b. (Brunton, pp 732-733; Katzung, pp 183-185, 219-220, 299, 1042t.) ACE inhibitors (and angiotensin receptor blockers, ARBs, eg, losartan) are contraindicated in pregnancy (category X), and should not be administered to “women of childbearing potential”—not just women who are pregnant—in the first place. Normal in utero development of the kidneys and other urogenital structures seems to be angiotensin-dependent. ACE inhibitors, or ARBs, given during the second or third trimesters (not the first) have been associated with severe and sometimes fatal developmental anomalies of these structures. Cranial hypoplasia, and neonatal hyperkalemia and hypotension, have also been reported.
α-Methyldopa (a) is considered one of the preferred antihypertensive drugs for pregnant women. It not only controls maternal blood pressure well, but also is remarkably free of adverse effects on the fetus. (Indeed, it is the most likely drug that we would prescribe for this hypertensive and now pregnant woman.)
β-Blockers such as labetalol (d) are sometimes used during pregnancy, posing no specific or significant risks to the mother or the fetus, provided that adequate perinatal care is given and blood pressure doesn’t fall excessively. None of the calcium channel blockers (including verapamil, e) seem to have any significant benefits or risks during pregnancy, compared with other drugs. (Verapamil, for example, is in pregnancy category C.) One theoretical concern with verapamil and diltiazem is a prolongation of labor as parturition draws near, due to suppression of uterine contractility.
There are concerns with using diuretics during pregnancy. Furosemide (c) or other loop diuretics pose relatively significant fetal risks. However, that is related to potential maternal hypovolemia and hypotension that may lead to placental underperfusion. There is no teratogenic or embryopathic risk on par with that associated with the angiotensin modifiers (ACE inhibitors or ARBs). Moreover, when the goal is treating essential hypertension in the absence of hypervolemia, edema, ascites, and so on, loop diuretics seldom are used. Instead, thiazide or thiazide-like diuretics are the ones usually chosen. Nevertheless, the thiazides also pose a risk of placental underperfusion, and so they are not preferred or ideal antihypertensives for the pregnant woman.
Note: It is not necessary (now) for you to memorize the pregnancy classifications of the nonteratogenic drugs listed above. What you need to know is that ACE inhibitors and ARBs are absolutely contraindicated. So is warfarin, at least during the first 12 weeks of gestation, which is addressed in Question 199.
195. The answer is c. (Brunton, pp 868-870; Katzung, pp 613, 617.) Clopidogrel (and the much lesser-used drug ticlopidine) is a noncompetitive antagonist of ADP. This prodrug (it must be metabolically activated) causes largely irreversible (ie, for the lifetime of the platelet) inhibition of platelet aggregation by blocking ADP binding to the Gi coupled P2Y(AC) receptor. It has no effect on platelet activation and amplification caused by such other proaggregatory agonists as collagen, thromboxane A2, thrombin, PAF, serotonin, or epinephrine.
Aspirin (a) inhibits platelet aggregation caused by thromboxane A2 (TXA2) only, and does so only by inhibiting TXA2 synthesis via cyclooxygenase, not by blocking TXA2 receptors.
Bivalirudin (b) is a synthetic hirudin derivative (you may recall that hirudin is produced by the medicinal leach, Hirudo medicinalis). It is classified as an anticoagulant, not as an antiplatelet drug. It is a direct-acting inhibitor of free and clot-bound thrombin, which leads to two main effects: (1) decreased conversion of fibrinogen to fibrin; and (2) reduced activation of Factor XIIIa, which in turn decreases conversion of soluble fibrin monomers to insoluble (polymerized) fibrin.
Bivalirudin is given IV as an alternative to heparin (both drugs bind to free thrombin, but bivalirudin also interacts with clot-bound thrombin), mainly along with aspirin for patients with unstable angina who are undergoing angioplasty. It is also used to help treat heparin-induced thrombocytopenia; and seems to be more effective than heparin when given postmyocardial infarction. A related drug is argatroban.
Heparin and warfarin (d, e) have no direct effects on platelets. (You should also recall that since warfarin’s site of action is the liver, it has no anticoagulant effects when tested in vitro (ie, added to a tube of whole blood).
Note: I mentioned that ticlopidine is much lesser-used than clopidogrel. Actually, it’s seldom used. The reason? A greater risk of thrombotic thrombocytopenic purpura (TTP) and/or neutropenia.
196. The answer is e. (Brunton, pp 765-767; Katzung, pp 193-208.) The vascular calcium channel-blocking actions of verapamil or diltiazem (nondihydropyridine class) will not only lower systemic blood pressure, but also tend to counter coronary arterial calcium influx that favors vasospasm. Thus, in this setting we can expect both antihypertensive and antianginal effects from one drug.
One might argue that nifedipine (answer c) would be a reasonable alternative. However, recall that the dihydropyridines (which is the class to which nifedipine belongs) lack cardiac depressant actions. Rapidly acting formulations of a dihydropyridine, even if given orally, are likely to lower blood pressure (the desired antihypertensive effect), but lacking cardiac-depressant actions are liable to trigger reflex (sympathetic/baroreceptor) cardiac stimulation. The resulting increases of either or both cardiac rate and contractility (mediated, reflexly, by sympathetic activation) necessarily may raise myocardial oxygen demand sufficient to cause myocardial ischemia, and/or may trigger coronary vasospasm. (The degree to which a dihydropyridine will cause unwanted reflex cardiac stimulation depends on the drug, the dose, and even the dosage form [eg, immediate-acting vs extended-acting].) Nonetheless, a nondihydropyridine would have a much better overall profile of cardiac/vascular actions.
ACE inhibitors or an ARB (a), or a thiazide (d), might cause no particular problems for a patient with vasospastic angina, but they also have no actions that would help that comorbidity. Why select one of them when a nondihydropyridine calcium channel blocker might be beneficial for both problems?
β-Blockers (b) would be a poor choice. Recall that the coronary vasculature constricts in response to α-adrenergic influences, while vascular β2 receptor activation tends to cause coronary vasodilation. Patients with vasospastic angina depend on β-mediated vasodilatory influences, and are at greater risk of ischemic events if the β-receptors are blocked.
197. The answer is c. (Brunton, pp 899-901, 972; Katzung, pp 627-628.) Answering this question correctly indeed depends on your knowledge of several drugs, and their prototype or representative agents, that goes beyond “cardiovascular” (since I included a diabetes drug [metformin] and an antidepressant [escitalopram] as possible answer choices).
Nonetheless, niacin (nicotinic acid; vitamin B3) is often added-on—in dosages higher than those recommended for vitamin supplementation—mainly to lower HDL and triglyceride levels, particularly in patients with the classic mixed hyperlipidemia characterized by elevated cholesterol and triglyceride levels (Type IIb) and who aren’t adequately managed with a statin. The primary effect of niacin is to decrease production of VLDLs (mainly by inhibiting lipolysis in adipocytes). Since LDLs are by-products of VLDL degradation, the fall in VLDL levels causes LDL levels to fall as well. Lastly, recall that VLDLs deliver triglycerides to and increase their storage in adipose tissue. By decreasing VLDL production, then, niacin indirectly counters triglyceride accumulation and favors triglyceride clearance (elimination). See the answer for Question 258 for more information about niacin’s more common side effects.
Pravastatin and other statins (HMG CoA reductase inhibitors), while mainly used for hypercholesterolemia (to lower LDL-cholesterol levels), often concomitantly raise HDL and lower triglyceride levels adequately. When statins and lifestyle modifications fail to control triglyceride levels adequately, adding niacin is usually a reasonable next step.
Citalopram (a), escitalopram, and other SSRI-antidepressants have no known effects on altered vitamin absorption or on the cardiovascular system. Metformin (b), the prototype biguanide often used for managing type 2 diabetes mellitus, typically suppresses (not increases) appetite. Statins do not cause peripheral neuropathy (d); and ACE inhibitors (ramipril and others; e) do not cause diabetic nephropathy, although they are clearly contraindicated for patients with severe bilateral renal arterial stenosis, such as that associated with severe and poorly controlled diabetes mellitus.
198. The answer is d. (Brunton, pp 828, 844, 1405-1407; Katzung, pp 238, 921-922.) Quinidine is becoming an outmoded drug, but it is, of course, still being used. It may be described as a broad-spectrum antiarrhythmic (that does not mean it is indicated or safe for all arrhythmias and all etiologies). It is mainly used for long-term management of supraventricular and ventricular arrhythmias: atrial flutter and fibrillation; and sustained ventricular tachycardia. The drug’s effects reflect direct ion channel-blocking actions plus a variety of receptor-blocking effects that may counteract the direct effects.
Direct blockade of sodium channels (a “major” action of Class I antiarrhythmics) slows impulse conduction in the atria, ventricles, and His-Purkinje system. Repolarization at those sites tends to be delayed, presumably by blockade of potassium channels. Among other changes, the ECG shows widened QRS complexes and prolonged QT intervals (which may predispose patients, particularly those with long QT syndrome, to some serious arrhythmias, including torsades).
Quinidine has strong anticholinergic (vagolytic) effects that are particularly apparent at the SA node (increased automaticity, tends to predominate over direct depressant effects from sodium channel blockade) and the AV node (increased conduction velocity, correct answer d). (Recall that vagal tone on the SA node slows spontaneous depolarization and heart rate; the vagolytic effects of quinidine will therefore do the opposite.) The effects on AV conduction are a main reason why, when quinidine therapy is to be started and atrial rates are still high, we pretreat the patient with a dose of a drug that “blocks down” the AV node: digoxin, sometimes verapamil, and occasionally a β-blocker. The main reason why ventricular rates aren’t identical (or close to) atrial rates during atrial fibrillation is because the AV node cannot transmit impulses at such high rates. This protective effect depends on AV nodal refractoriness and a relative inability to transmit too many impulses per time. If we did not suppress the AV node before giving quinidine, the AV nodal “conduction-stimulating” effects of the quinidine might increase AV nodal transmission; ventricular rates might rise to dangerous levels as the atrial rate slows in response to the quinidine.
Quinidine is not likely to increase blood pressure (b), or worsen preexisting hypertension. Quite the contrary: the predominant vascular effect of the drug is dilation, and blood pressure falls, due to peripheral vascular α-adrenergic blocking activity. Likewise, quinidine exerts a negative inotropic effect—not a positive one (e)—on ventricular muscle. That, too, can contribute to a fall of blood pressure. For long-term management of a patient with atrial fibrillation, anticoagulants are important (not contraindicated, c) for prophylaxis of venous thrombosis; the use of quinidine does not contraindicate that.
199. The answer is e. (Brunton, pp 857, 865; Katzung, pp 608-611, 1043t.) The scenario describes the classic findings with “warfarin embryopathy”: nasal deformities (mainly a flattening of the nose) and stippling (based on standard x-ray images) at the epiphyses of the long bones. The drug is, as noted, absolutely contraindicated during pregnancy. The fetal risks are greatest if warfarin is taken at any time during the first 12 or so weeks of gestation, but adverse fetal effects can occur later. They are irreversible, sometimes fatal; there is no drug or vitamin (eg, vitamin K, folate) therapy that will be of benefit. All the other drugs listed pose some pregnancy-related risks (but lower and with less supporting data than with warfarin), and they do not share the characteristics of warfarin embryopathy. Clonidine (a) is in pregnancy category C. Its antihypertensive mechanism is largely identical to that of methyldopa, which is in category B and usually is a preferred drug for hypertension during pregnancy. Low molecular weight heparin (b) is also in pregnancy category B. If anticoagulation is required during pregnancy, it is the drug that most likely will be used. Hydrochlorothiazide (or thiazide diuretics in general) has no known human teratogenic effects (category B). Nitroglycerin is not contraindicated because of teratogenesis (category C).
Note: I’ll kindly ask you to review the answers and explanations for Question 194, and recall that even though a cardiovascular drug may not be teratogenic nor absolutely contraindicated in pregnancy, it can cause adverse (potentially fatal) fetal effects if, for example, it causes hypotension and/or hypovolemia, which can significantly reduce placental blood flow. Recall that warfarin, ACE inhibitors, and angiotensin receptor blockers are contraindicated in pregnancy.
200. The answer is d. (Brunton, pp 297, 780-781; Katzung, pp 175-176.) Among all the common oral antihypertensives, a Coombs-positive test is associated only with methyldopa. It occurs in up to about 20% of patients taking this drug long term. It may progress to hemolytic anemia, but that seldom occurs. The probable cause is an immune reaction (IgG antibodies) directed against and potentially lysing the red cell membrane. Other drugs with the potential to cause an immunohemolytic anemia are penicillins, quinidine, procainamide, and sulfonamides.
201. The answer is a. (Brunton, pp 206t, 306f, 309, 774; Katzung, pp 151-152, 154-156.) Phenoxybenzamine is a long-acting and noncompetitive α-adrenergic blocker. It works by alkylating (rather than merely occupying and “blocking”) α-adrenergic receptors, thereby rendering them incapable of interacting with α-adrenergic agonists such as epinephrine (which is being released in abundance by the pheochromocytoma). The drug has no ability to block β-adrenergic receptors (b), inhibit catecholamine synthesis (c), inhibit angiotensin converting enzyme (d), or affect any of the major enzymes involved in catecholamine degradation (COMT, answer e; or MAO).
202. The answer is c. (Brunton, pp 328-329; Katzung, pp 157-161, 165-166.) No β-blocker ordinarily should be administered to patients with asthma, because of a great risk of severe and potentially fatal bronchoconstriction or bronchospasm (unless the β-blocker is being given for a medical emergency that requires β-blockade, and likely benefits outweigh likely risks). This applies to all classes of β-blockers: nonselective, like propranolol; β1 “selective,” that is, atenolol or metoprolol; those with intrinsic sympathomimetic/partial agonist activity, such as pindolol; and those that also have α-blocking activity, that is, labetalol and carvedilol. The contraindication applies to all administration routes, including topical (ophthalmic); there are clinical reports of fatal bronchospasm induced by topical ophthalmic administration of “just one drop” of β-blockers.
The reason? Airways of persons with asthma are exquisitely sensitive to a host of bronchoconstrictor stimuli and exquisitely dependent on the bronchodilator effects of circulating epinephrine (β2-receptors). Block those β-receptors, and that sets the stage for bronchoconstriction or bronchospasm. Another important factor is that this patient needs albuterol for occasional relief of bronchospasm. The drug works, of course, by activating β2 adrenergic receptors in the airways. Any or all of the available β-blockers will antagonize albuterol’s desired effects, the so-called cardioselective agents included, because their selectivity for β1-receptors is only relative, not absolute; and it is highly dose-dependent.
Diltiazem (a) and particularly verapamil (e), both nondihydropyridine calcium channel blockers, might be a good choice for this patient. They not only lower blood pressure by blocking calcium influx into vascular smooth muscle, but also (theoretically, at least) do the same in airway smooth muscle, thereby preventing or at least reducing bronchoconstriction. Hydrochlorothiazide possibly could exacerbate the problems by causing excessive fluid loss (which could favor bronchoconstriction), but the chances of this are low. (Loop diuretics such as furosemide would be of more concern, owing to their greater efficacy in terms of causing loss of circulating fluid volume). There are no specific concerns with using ramipril, an angiotensin converting enzyme inhibitor. (Remember: Drugs with generic names ending in “-pril” are ACE inhibitors, so even if you learned about only captopril as an ACE inhibitor, you should have realized that ramipril is in the same class.)
Note: It’s fair to say that the “Big 4” classes of antihypertensive drugs can be summarized as A, B, C, D: ACE inhibitors or angiotensin receptor blockers; Beta-blockers; Calcium channel blockers; and Diuretics (thiazides or thiazide-like agents such as metolazone). Which is “best” for a particular patient depends on a host of patient-related factors that may weigh in favor of selecting a drug from a particular class (A, B, C, D) or weigh against a particular class. There has been a resurgence in the use and recommendation of thiazides or thiazide-like drugs for initiating anti-hypertensive therapy. Likewise, much more recently, and for a variety of reasons, some experts are stating that β-blockers should be dropped from the Big 4 list for most patients.
203. The answer is d. (Brunton, pp 801-804, 829t, 835t, 837-839; Katzung, pp 215-218, 221, 1036-1037.) Of the effects listed here, you would expect to find slowed AV nodal conduction velocity in response to digoxin. This is a common and, in many situations useful, effect. For example, when we give the drug as part of the pharmacologic management of atrial fibrillation or flutter, the main desired response is not suppression of the arrhythmia per se, but rather to “block down” the AV node so that as atrial rates fall (but are still high), the AV node will be unable to transmit the same frequency of impulses to the ventricles. That is, we “protect” the ventricles from excessive acceleration by inducing a degree of AV block. In terms of more specific effects on the AV node, digoxin slows conduction velocity and increases AV nodal refractory periods.
Other “predominant” effects on the heart—all concentration-dependent—include increases of both atrial and ventricular automaticity and of conduction velocity through those structures and the His-Purkinje system.
204. The answer is d. (Brunton, pp 767t, 779-781; Katzung, pp 181, 189, 220.) Hydralazine predominately dilates arterioles, with negligible effects on venous capacitance. It typically lowers blood pressure “so well” and quickly that it can trigger two unwanted cardiovascular responses that usually need to be dealt with:
1. Reflex cardiac stimulation (involving the baroreceptor reflex) is common, and it is typically managed with a β-adrenergic blocker (unless it is contraindicated). An alternative approach would be to use either verapamil or diltiazem (but not a dihydropyridine-type calcium channel blocker such as nifedipine, which would not suppress—and, in fact might aggravate—the reflex cardiac stimulation).
2. Activation of the renin-angiotensin-aldosterone system to compensate for what the body believes is an excessive and unwanted fall of blood pressure. One consequence of this unwanted response is increased renal sodium retention that would expand circulating fluid volume and counteract hydralazine’s pressure-lowering effects. This is typically managed with a diuretic. A thiazide may be sufficient to combat the sodium retention, but a more efficacious diuretic (loop diuretic) may be necessary and is usually selected.
Captopril (or another ACE inhibitor, or an angiotensin receptor blocker such as losartan) might be a suitable add-on (it would cause synergistic antihypertensive effects and prevent aldosterone-mediated renal effects). However, combining it with nifedipine (dihydropyridine calcium channel blocker) is irrational. As noted above, given the “pure” vasodilator actions of nifedipine and the lack of cardiac-depressing activity (as occurs with verapamil), the net effect on heart rate would be either no suppression of the tachycardia or a worsening of it.
Digoxin, alone or with virtually any other drug (c), is not rational because it would not meet the goals of blunting reflex cardiac stimulation and efficiently counteracting the consequences of renin-angiotensin-aldosterone system activation. There is no indication that there is need for inotropic support in this patient.
Spironolactone, alone or with digoxin, would be of little benefit. One could argue that by virtue of the spironolactone’s ability to induce diuresis by blocking aldosterone’s renal tubular effects, it would counteract hydralazine’s ability to lead to renal sodium retention. That may be true, but spironolactone (with or without digoxin) will do nothing desirable to blunt the unwanted reflex cardiac activation.
Nitroglycerin (e) would add to hydralazine’s antihypertensive effects, but it would probably aggravate (and certainly not counteract) the reflex cardiac stimulation, and may also increase the unwanted renal response (via a hemodynamic mechanism).
Both triamterene and amiloride are potassium-sparing diuretics. The combination of two diuretics in this class is almost always irrational (it can lead to hyperkalemia). Either might beneficially combat a propensity for renal sodium retention in response to hydralazine. But, as with any diuretic alone, either or both would do little if anything to control the unwanted cardiac response.
Vitamin K (included in answer c) was included as a foil. If you are associating hydralazine with some vitamin-related problem, you should be thinking of vitamin B6 (pyridoxine): hydralazine can interfere with B6 metabolism, causing such symptoms as peripheral neuritis, and so prophylactic B6 supplementation is often used along with long-term hydralazine therapy. Hydralazine is also known to cause a lupus-like syndrome. While prophylaxis with pyridoxine helps with the potential neuritis it does nothing for the lupus-like syndrome.
Note: Nowadays many physicians consider hydralazine to be (at best) a fifth or sixth line agent for chronic hypertension. Regardless, the drug is not a “go to” drug unless, essentially, all other reasonable options have been tried and shown to be ineffective.
205. The answer is a. (Brunton, p 725; Katzung, pp 295-300.) The bradycardia caused by the unknown drug is reflex-mediated by baroreceptor activation in response to the rise of blood pressure. Angiotensin II, by activating vascular A-II receptors, raises blood pressure and peripheral resistance, and that response would not be inhibited by pretreatment with prazosin (selective α1-adrenergic antagonist), nor any other adrenergic blocker (antagonist) for that matter.
The bradycardia, of course, involves reflex parasympathetic activation (and relative and simultaneous withdrawal of sympathetic tone to the heart), release of ACh from the vagus and activation of muscarinic receptors on the SA node. Atropine pretreatment indeed prevents that response, as described in the question—and regardless of whether the original pressor response was caused by A-II, or any other vasopressor drug for that matter.
Dobutamine (b) doesn’t fit the bill; it is largely a selective β1 agonist. For all practical purposes it causes no vasoconstriction (an α1 response if we limit things to adrenergic drugs), and via the β1 activation will directly increase heart rate, contractility, automaticity, and electrical impulse conduction (eg, through the AV node). No α-activation occurs (rises of blood pressure, if they occur in response to dobutamine, are due to the positive inotropic effect), and so prazosin would have no direct effect on responses to dobutamine. Isoproterenol (c) can be ruled-out for largely similar reasons; it activates both β1 and β2 receptors; and lacks vasoconstrictor activity, whether due to activation of α-adrenergic receptors or by other mechanisms.
Could norepinephrine (d) be a reasonable answer? It certainly causes a vasopressor response and reflex bradycardia (at usual doses). However, the question stated that the unknown drug’s pressor response is not inhibited by α-blockade. Since a major element of NE’s pressor effect depends on α activation, it’s not a reasonable choice. The same applies to phenylephrine (e): this nonselective α-agonist also causes a pressor response that would be reduced or blocked altogether by prazosin pretreatment.
206. The answer is d. (Brunton, pp 390, 793; Katzung, pp 182, 189, 334.) Cyanide is the ultimate toxic metabolite of nitroprusside sodium. The drug is initially metabolically reduced to nitric oxide, which is responsible for the arteriolar and venular dilation. However, it is another metabolite, CN– (and to a lesser extent the next metabolite, thiocyanate) that is the main cause of or contributor to toxicity.
When CN– accumulates to sufficiently high levels (as from excessive or excessively prolonged administration of the drug), the vasculature develops what amounts to a tolerance to the drug’s vasodilator effects, and so blood pressure usually starts to rise despite the presence of high drug levels. Toxic cyanide accumulation can also lead to severe lactic acidosis: the CN– reacts with Fe3+ in mitochondrial cytochrome oxidase, inhibiting oxidative phosphorylation. Other characteristic signs and symptoms of the toxic syndrome include a cherry red skin (because mitochondrial oxygen consumption is blocked, venous blood remains oxygenated and as “bright red” as normal arterial blood), hypoxia, and, ultimately, hypoxic seizures and ventilatory arrest. Note: Nitroglycerin and other similar nitrovasodilators are not metabolized to CN– and so manifestations of CN– poisoning are not elements of their toxicity profile.
207. The answer is c. (Brunton, pp 682-684, 777, 790-791; Katzung, pp 189, 201, 334.) Hypokalemia due to the effects of potassium-wasting diuretics such as furosemide increase susceptibility to digoxin toxicity, and they are probably the most common cause of it. How does this occur? Digoxin binds to a K+-binding site on the sodium pump (ATPase). That is, there is competition between digoxin molecules and K+ for the same binding sites. When potassium levels are low (as can occur with any potassium-wasting diuretic, ie, a loop, thiazide, or thiazide-like diuretic), more digoxin molecules are able to bind to the ATPase—even though the actual digoxin concentration in the bloodstream hasn’t changed. More binding leads to greater ATPase inhibition: an intensified digoxin effect that, quite often, can lead to adverse responses rather than better therapeutic effects.
Captopril (a) has no direct effects on digoxin’s actions. (We might add that an ACE inhibitor such as captopril, along with a β-blocker and a diuretic—not digoxin—are now considered the preferred therapy for most patients with recent onset and mild heart failure.) Cholestyramine (b), a cholesterol-binding resin, interacts with concomitantly administered (oral) digoxin to reduce digoxin absorption. It would not increase the risk of digoxin toxicity; quite the opposite, it would reduce digoxin’s therapeutic effectiveness. Lovastatin (d; an HMG-Co-A reductase inhibitor/“statin”) and nitroglycerin (e) are not likely to cause the observed toxicity either.
208. The answer is c. (Brunton, pp 830, 831, 764-765; Katzung, pp 202t, 238t.) The etiology involves coronary vasospasm, and that can be blocked well with a calcium channel blocker (CCB): diltiazem, verapamil, or dihydropyridines (eg, nifedipine, for which slow-/extended-acting oral formulations are used). The CCBs block coronary vascular smooth muscle influx of calcium, which is a critical process in triggering vasospasm.
Nitroglycerin seems to be marginally effective in terms of long-term symptom relief, although it may be the only rapidly acting drug that will be efficacious for acute angina and self-medication.
Aspirin, through its antiplatelet aggregatory effects, would be beneficial, prophylactically, if coronary thrombosis were part of the etiology of variant angina, but thrombosis is not the main problem with coronary vasospasm.
Atorvastatin (or other statins) are useful for primary prevention of coronary heart disease, but coronaries that undergo spasm may be remarkably free of atherosclerotic plaque, and the statins have no antispasmodic effects per se.
The β-blockers, which are important drugs for many patients with ischemic heart disease, can do more harm than good in vasospastic angina. In essence, β-receptor activation in the coronaries tends to cause vasodilation, an effect that to a degree counteracts simultaneous α-mediated constriction. Block only the β-receptors and the α-mediated constrictor effects—vasospasm-favoring effects—are left unopposed. Variant angina, then, is likely to be made worse, not better, with β-blockers. (You might ask whether a combined α-/β-blocker like labetalol or carvedilol might be better than a nonselective or cardioselective β-blocker. Perhaps in theory, but not in practice. Remember that the α-blocking effects of these drugs are comparatively weak; their β-blocking, spasm-favoring effects will predominate.)
209. The answer is c. (Brunton, pp 821t, 1405-1407; Katzung, pp 238, 921-922, 1160t.) Digoxin toxicity is likely to occur within 24 to 48 hours unless the digoxin dose is adjusted down. The reason is that quinidine will reduce the renal excretion of digoxin (digoxin’s main elimination route). This is probably due to some mechanism by which quinidine inhibits P-glycoprotein transport of digoxin in the kidneys. (See the answer for question 10, on page 28, for a short table listing some drugs that are substrates, inducers, and inhibitors of P-glycoprotein.)
There is no “reverse interaction”—that is, an ability of digoxin to cause signs and symptoms of quinidine toxicity (a). Quinidine has no significant impact on the renal actions of any diuretics, whether these actions are expressed in terms of urine output (volume or concentration) or renal handling of sodium or potassium or other electrolytes or solutes (b, d).
Quinidine-induced digoxin toxicity may suppress cardiac contractility, but that would not necessarily be a direct effect of an interaction on the inotropic state of the myocardium. Rather, it would be secondary to potential digoxin-induced arrhythmias, and it would not occur “promptly.”
Quinidine does cause some drug-drug interactions by pharmacokinetic mechanisms. It is a potent inhibitor of CYP2D6, and can (for example) inhibit the analgesic effects of codeine by inhibiting its metabolism to morphine. However, this mechanism does not apply to the quinidinedigoxin interaction; digoxin is eliminated completely by the kidneys, with no prior metabolism.
210. The answer is e. (Brunton, pp 828, 835t, 840-841; Katzung, pp 241-242, 246-248.) The class I-C antiarrhythmics are associated with a higher incidence of severe proarrhythmic events than virtually any other antiarrhythmics in other classes. This risk partially explains why, when these drugs were first approved, they were indicated only for life-threatening ventricular arrhythmias that failed to respond to all other reasonable (and safer) alternatives. (This risk also contributed to why another I-C agent, encainide, was withdrawn from the market.)
Nowadays, these I-C agents are still used for serious (life-threatening) and refractory ventricular arrhythmias, their efficacy arising from significant sodium channel blockade. However, they also block some potassium channels, which accounts for modestly growing interest in and use of these drugs for some atrial arrhythmias. Regardless of whether the use is for an atrial or ventricular arrhythmia, the proarrhythmic effects should be of concern.
Pulmonary fibrosis and alterations of thyroid hormone status (typically, a hypothyroid-like state) are uniquely associated with amiodarone (among all the antiarrhythmics), and amiodarone was not one of the answer choices.
Note: I’ll opine that memorizing which antiarrhythmic agents are in which Vaughn-Williams class may not be profitable (except for answering nit-picky and clinically irrelevant exam questions). Among the reasons why (1) this classification is based largely on electrophysiologic effects of the drugs in largely normal, isolated cardiac cells, not in diseased intact human hearts; (2) some antiarrhythmic drugs have electrophysiologic/ionic mechanisms of action that would reasonably place them in more than one Vaughn-Williams class (eg, amiodarone); (3) belonging to a particular Vaughn-Williams class does not necessarily predict clinical use of the antiarrhythmic; and (4) side effects profiles and toxicities of drugs in the same Vaughn-Williams class—both cardiac and extracardiac—can differ substantially.
211. The answer is b. (Brunton, pp 752, 766-767, 830-831; Katzung, pp 183, 194, 201-205, 245.) In a nutshell, this question summarizes some of the main and important differences between the dihydropyridine CCBs (eg, nifedipine, and especially in immediate-release formulations, plus many others) and the nondihydropyridines (particularly verapamil, less so with diltiazem). The dihydropyridines are quite “selective” for their vascular effects. They cause significant arteriolar dilation, which usually activates the baroreceptor reflex that increases sympathetic influences on the heart: positive inotropy, positive chronotropy (reflex tachycardia can be severe), and increased automaticity and conduction velocity (dromotropic effects).
In contrast, and importantly, verapamil and diltiazem cause not only arteriolar dilation (via calcium channel blockade in vascular smooth muscle), but also direct cardiac depressant effects (due to calcium channel blockade in cardiac tissues). These cardiac effects oppose, or blunt, reflex sympathetic cardiac activation that otherwise would occur in response to only peripheral vasodilation: such problems as reflex tachycardia and positive inotropy are much less—often nonexistent—with the nondihydropyridines. In fact, when reflex cardiac stimulation caused by other drugs (eg, nitroglycerin) is problematic and must be controlled, either vera-pamil (probably preferred) or diltiazem may be a reasonable alternative to the traditional agents for blocking the unwanted cardiac responses: the β-adrenergic blockers. Moreover, a nondihydropyridine CCB is usually the drug of choice for controlling cardiac stimulatory responses when a β-blocker is contraindicated (eg, in asthma). Be aware that combined use of diltiazem or verapamil with a β-blocker is risky due to possibility of additive or greater inhibitory effects on the cardiac rate, contractility, and especially AV nodal function. And, whether used alone or in combination, β-blockers and the nondihydropyridine CCBs (verapamil, diltiazem) should not be administered to patients with severe heart failure due to the added risks from further suppression of cardiac output.
Notes: The vasodilator effects of verapamil are weaker than those of diltiazem, but the peripheral vascular (vasodilating) effects of either are still far weaker than those of a dihydropyridine. Diltiazem is classified, chemically, as a benzothiazepine (similar to but not to be confused with benzodiazepines—diazepam, midazolam, and many other drugs used for their CNS effects.) Verapamil is, chemically, a phenylalkylamine. However, memorizing the names of these chemical groups is probably a waste of your time: these drugs are almost always referred to simply as nondihydropyridines.
212. The answer is b. (Brunton, pp 86t, 390, 793; Katzung, pp 182, 189, 334.) Nitroprusside’s main toxic metabolite is cyanide. Nowadays, a preferred and effective approach to managing cyanide toxicity from nitroprus-side overdose is to administer hydroxycobalamin. It reacts with cyanide, forming cyanocobalamin, which is largely nontoxic (and is considered a form of vitamin B12).
A more traditional (older) approach to managing cyanide toxicity—one that is still occasionally used, is based on the fact that cyanide normally is metabolized to the less toxic and more readily excreted thiocyanate by an enzymatic process (rhodanese is the key enzyme) that uses thiosulfate as a cofactor. Endogenous thiosulfate stores are rather limited, and with more serious degrees of cyanide (or nitroprusside) toxicity the thiosulfate is readily depleted. Other endogenous sulfur-containing substrates are depleted too. This traditional treatment plan, therefore, involves administering sodium thiosulfate to restore thiocyanate production. Nitrites (amyl and sodium nitrites) are administered to induce methemoglobinemia. The methemoglobin then binds free cyanide, forming less toxic cyan(o) methemoglobin.
None of the other drugs listed play a role in the management of cyanide poisoning, whether from nitroprusside excess or from other causes (eg, suicide attempts, environmental exposure to cyanide). Aminocaproic acid (a) is an antithrombolytic (antifibrinolytic) drug used, for example, when such drugs as alteplase (t-PA; plasminogen activators) cause excessive thrombolysis that needs to be counteracted pharmacologically. The drug inhibits the actions of not only therapeutic plasminogen activators, but also those of endogenous activators. It also weakly inhibits plasmin activity. Protamine sulfate (c) should be remembered as the antidote for excessive anticoagulation caused by heparin (which occurs mainly with unfractionated, rather than low molecular weight, heparin). Thrombin (d) is a prothrombotic agent that plays no role in nitroprusside (or cyanide) toxicity. Vitamin K (e) is used to antagonize excessive effects of warfarin.
213. The answer is a. (Brunton, pp 730-736, 753, 784-785; Katzung, pp 170-174, 185-188.) Although combined use of an ACE inhibitor (or angiotensin receptor blocker [ARB], eg, losartan) and a diuretic is quite common, great care must be taken when adding one of the drugs to therapy that has been started with the other. The reason is that some patients develop a sudden fall of blood pressure that may be sufficient to cause syncope or other complications. Volume and sodium depletion seem to be among several probable causative factors.
Answer b is incorrect. The effects of ACE inhibitors (or ARBs) and thiazides on renal handling of potassium are the opposite of one another, not synergistic. ACE inhibitors tend to elevate potassium levels (in part, by lowering aldosterone levels); the thiazides (and loop diuretics, eg, furosemide) are potassium-wasting.
There is no evidence that adding one of these drugs to therapy with the other can cause acute (or chronic) heart failure (c); indeed, such a combination is often an essential component in managing chronic heart failure. Blood pressure will fall, not rise, and certainly not cause hypertensive crisis (d); and slowed AV nodal conduction rates (e) due to this drug combination do not occur.
214. The answer is d. (Brunton, pp 853-859; Katzung, pp 604-607.) Administration of protamine sulfate will quickly reverse the bleeding caused by excessive effects of heparin. Protamine binds to heparin, forming a stable complex that abolishes heparin’s anticoagulant activity. It may also have its own anticoagulant effect by binding with platelets and fibrinogen. Aminocaproic acid (a) binds avidly to plasmin and plasminogen, and so is an effective inhibitor of fibrinolysis and an antagonist of fibrinolytic/thrombolytic drugs. Dipyridamole weakly blocks platelet ADP receptors, and has a long but uninspiring history as an antiplatelet drug. Factor IX will not reverse the excessive effects of heparin. Vitamin K is used as an antidote for excessive effects of such drugs as warfarin, and does nothing for heparin overdoses.
215. The answer is c. (Brunton, pp 758-759; Katzung, p 199.) Nitroglycerin causes its vasodilator effects via a nitric oxide (NO)- and cyclic GMP-dependent mechanism. The NO activates guanylyl cyclase which forms cGMP from GMP. The cGMP, in turn, dephosphorylates myosin light chains, leading to reduced actin-myosin interactions and relaxation of the smooth muscle cells. Normally, this vascular effect is modulated by cGMP degradation (to GMP) by cGMP-specific phosphodiesterases (PDEs). Sildenafil (and the related drugs tadalafil and vardenafil) inhibit the activity of those PDEs, thereby maintaining cGMP levels and potentiating the vasodilator effects. This interaction may lead to severe excessive effects, including life-threatening hypotension and myocardial and cerebral ischemia. Sildenafil and related drugs also increase the risk of symptomatic hypotension if taken by people who are also being treated with α-adrenergic blockers, including those that selectively block α1 receptors (eg, prazosin and doxazosin) and are used for managing hypertension.
Note the important mechanistic links between vasodilation/hypotension, sexual intercourse, and potentially fatal cardiac responses. Sexual arousal—and, especially orgasm—causes a massive activation of the sympathetic nervous system. One consequence of that, α-mediated vasoconstriction that tends to keep blood pressure up, is too feeble to overcome the hypotensive effects of the sildenafil-nitroglycerin combination. Along with a fall of blood pressure is a fall of coronary perfusion pressure (diastolic blood pressure), that is, reduced myocardial blood flow/oxygen supply. Yet the sympathetic activation concomitantly causes significant increases of cardiac rate and contractility, that is, increased myocardial oxygen demand. Oxygen demand rises, supply falls, and the stage is set for acute myocardial ischemia.
A final point to consider. It is reasonable to assume that if the patient is taking any or even several of the drugs listed in the question, he has underlying cardiac disease. However, and in contrast with what some students have suggested, underlying cardiac disease per se does not automatically contraindicate or even excessively increase the risks from sexual intercourse and orgasm. (How severe is the patient’s cardiovascular disease? It is well controlled? What are the specific diseases and risks he has?) Similarly, none of the drugs listed other than the nitrovasodilators (and some α-blockers mentioned in the previous paragraph) contraindicate the use of any of the ED drugs.
216. The answer is e. (Brunton, pp 818-819, 823; Katzung, pp 233-235.) Although you may not consider this question a pharmacology question, being able to answer it correctly is important to the rational and safe use of many drugs.
In general, heart block refers to excessively slowed AV nodal conduction, which you assess by measuring the PR interval on the ECG. Recall that this interval gives information on how long it takes for electrical impulses that originate with SA nodal depolarization to pass through the atrial myocardia and the AV node, ultimately leading to ventricular activation (manifest as the QRS complex). Impulse conduction through the atria is normally quick; it is the AV node that has the slowest intrinsic impulse conduction rate anywhere in the heart, and it is arguably the structure that is most susceptible to drug- (or disease-) induced changes of supraventricular conduction that lead to the diagnosis of AV block.
In first-degree heart block, the main manifestation is a prolonged PR interval, but each P wave is followed by a normally generated QRS complex. In second-degree heart block, the PR interval is prolonged and some P waves are not conducted through the AV node, and so are not followed by a QRS triggered by the prior atrial activation (eg, 2:1 block). In third-degree (complete) heart block, no P waves are conducted normally through the AV node, and ventricular activation is solely dependent on intrinsic automaticity of the ventricles (or conducting tissue therein).
Auscultation of the heart, the presence of blood pressures that are above or below what is generally regarded as normal, or the presence of a slow sinus rhythm, are not reliable indicators of heart block.
217. The answer is e. (Brunton, pp 83t, 765-767; Katzung, pp 201-205, 245.) Constipation is a fairly common, sometimes very bothersome, and occasionally dangerous response to verapamil (more so than with any other calcium channel blocker). It is a widely used drug. The mechanism underlying this side effect isn’t known for sure, but probably lies in vera-pamil’s rather unique ability to block calcium channels on longitudinal and sphincter smooth muscle cells in the GI tract. Regardless of whether we know the mechanism for sure, it is important to know that verapamil tends to cause constipation in more than a handful of patients. Statins (a), ACE inhibitors (b), β-blockers of any class or organic nitrovasodilators such as nitroglycerin (d) do not cause constipation as a side effect.
218. The answer is a. (Brunton, pp 779-781; Katzung, pp 181, 220.) Hydralazine’s primary vasodilator actions occur on arterioles, not on venules. The others have both arteriolar and venular dilator activity, thereby lowering both afterload and central venous pressure (venous capacitance; right atrial pressure). Hydralazine’s probable mechanism of action involves a nitric oxide (NO)-mediated process. (There is also some evidence that the drug opens voltage-gated potassium channels on vascular smooth muscle cells, causing muscle cell hyperpolarization and ultimate vasodilation.)
The other drugs listed cause vascular smooth muscle relaxation on both arterioles and venules: losartan (b), by blocking angiotensin II receptors; nifedipine (c) via calcium channel blockade; nitroglycerin (d) by NO as an intermediate; and prazosin (e) by selective α1-adrenergic blockade. Here is a short table listing the main sites where various drugs or drug classes exert their main or sole vasodilator actions.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


