Permeation and Metabolism
Outline
The Importance of Drug Concentration125
New Terminology132
“Drug-Like” Properties of Molecules133
Drug Absorption135
Drug Metabolism142
Oral Bioavailability150
Summary152
This chapter discusses how the body is a balanced in vivo system of inflow (absorption) and outflow (clearance) of drug and how prospective drug molecules require a minimal set of physico-chemical properties (referred to as “drug-like” defined by LogP, H2O solubility, pKa and molecular weight) to be able to cross biological membranes to gain entry into the body. Passive diffusion and transport processes control absorption while metabolism and excretion govern removal of drugs from the body; many of these can be estimated in in vitro assays. In addition, assays to assess the stability of molecule to destruction by hepatic enzymes, the ability of a molecule to inhibit hepatic enzyme function (this can be predictive of drug–drug interactions) and the propensity of a molecule to produce induction of live enzymes can also be assayed to optimize pharmacokinetic profiles. Finally, a discussion of oral bioavailability as a dual process of absorption followed by metabolism (first pass effect) is considered.
Keywords
ADME, antedrug, bioavailability, clearance, drug–drug interaction, enzyme induction, first pass effect, lipophilicity, LogP (LogD), mechanism-based inhibition permeation (Papp), prodrug.
The Importance of Drug Concentration
When testing drugs in vitro, the drug is confined to a known volume (i.e., well of a plate, test tube, etc.), therefore the concentration of the drug is known. This is critical since all measures of drug activity (i.e., potency, efficacy, affinity) are basically concentrations at which a defined drug effect is observed. Therefore, if a concentration of 1μM is seen to produce a 50% inhibition of activity, then the IC50 is taken to be 1μM; this is a characteristic value that would indicate a higher potency over another drug with an IC50 of 10μM and a lower potency than one with an IC50 of 0.1μM. The key to this type of system is accurate knowledge of the concentration. A useful way to view experiments is to define independent and dependent variables. Independent variables are what experimenters put into the experiment and what they are required to know; in this case, drug concentration. What comes out of an experiment is data in the form of dependent variables, i.e., what the system being observed does with the independent variable to produce a system value, i.e., potency. Independent variables have only random error whereas dependent variables have random plus system error.
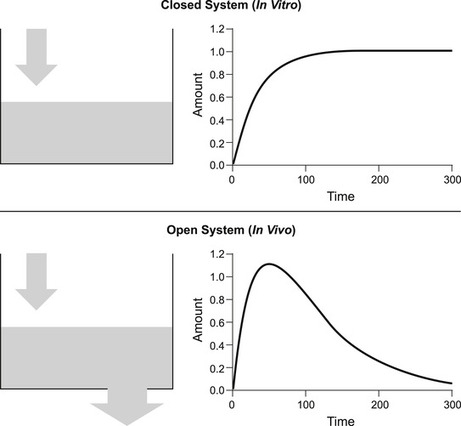
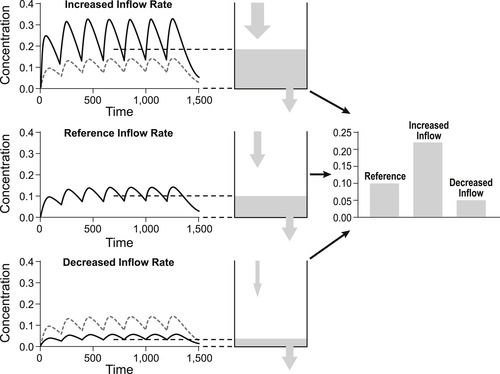
In vitro data describing potency is not usually associated with a time because measurements are taken after a steady-state or equilibrium has been attained. Figure 7.1 shows the concentration of drug added to a closed vessel; the kinetics show an increase with time to a steady-state after which the dependent variable attains a constant value, i.e., it is a closed system. In contrast, in vivo systems are like a vessel with a hole in it; equilibrium for a single dose is not attained and the level of fluid (e.g., concentration of drug) is very much dependent on the time that the measurement is taken, i.e., they are open systems (see Fig. 7.1). Pharmacokinetics is the science of accurately determining the concentration of drug in the body and devising methods of attaining steady-state levels of drug for therapy through repeat dosing. Figure 7.2 shows how giving repeated doses of a drug at regular intervals can achieve a steady-state level of drug in vivo. While a constant steady-state concentration of drug often can be attained in vivo with repeated dosing it should still be recognized that this is still a non-equilibrium system, and that the steady-state depends upon both the rate of entry and the rate of exit of the drug. Changes in either of these will subsequently alter the steady-state level of drug in the body. Figure 7.2 shows how an increase in the rate of entry of drug into the body leads to an increased steady-state, while a decrease in the rate of entry leads to a corresponding decrease in the steady-state. Figure 7.3 shows how the rate of exit of the drug correspondingly affects steady-state levels. Thus, an increased rate of exit leads to a reduced steady-state while a decrease in the rate of exit leads to an increased steady-state level. This chapter will discuss the various in vivo processes that control the rate of entry and exit of a drug from the body and the use of this knowledge to attain a therapeutically useful steady-state level.
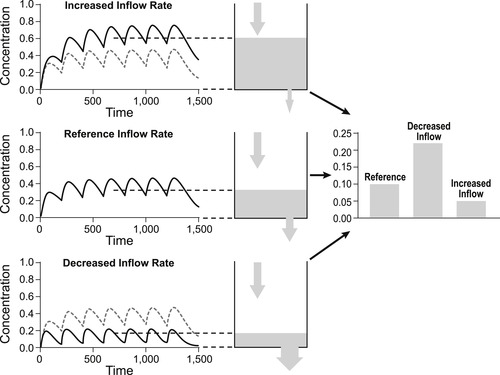 |
| Figure 7.3 As shown in Fig. 7.2, repeated dosing in an in vivo system can lead to the attainment of a steady-state that depends on the relative rates of entry and exit from the system. The top panel shows how a decreased rate of exit can lead to an increased steady-state level while the bottom panel shows how an increase in the rate of exit leads to a decrease in the steady-state level. |
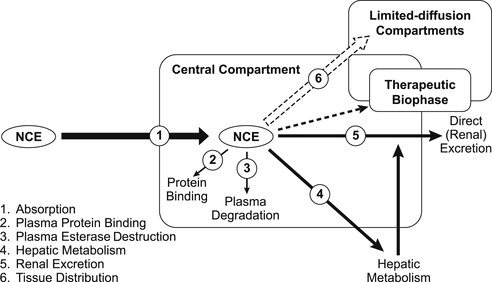
Figure 7.4 shows a schematic of the various processes that affect the entry into and exit out of the body. It is worth considering these separately as a prelude to understanding how they combine to affect in vivo drug concentration. An acronym (ADME) is often used to describe the interaction of a molecule with these processes: absorption; distribution; metabolism and excretion. Clearly, drugs require adequate ADME properties to be therapeutically useful; Box 7.1 shows how inferior pharmacokinetics can preclude otherwise favorable in vitro activity.
Box 7.1
Inadequate ADME properties can be devastating to otherwise good drug activity; shown below are two antitumor molecules, one five times more potent than the other. The poor pharmacokinetic properties of the more potent compound make it far less active in vivo. [7] Several iterations may be required before a suitable drug candidate profile is achieved. While the first histamine H2 antagonist for ulcer treatment, burimamide, had primary target activity, it required further work to develop metiamide (target active but an inadequate safety profile) and finally cimetidine (target active, safe and acceptable ADME properties) to achieve drug status. Pharmacokinetic problems led to a rate of 40% failure in drug development in 1991. Access to economical in vitro ADME assays led to a decrease in failure rate to 10% in 2000. [8]
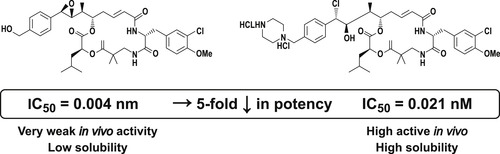 |
Drugs require three basic properties: primary activity at the therapeutic target; favorable ADME behavior to enter and stay in the body to produce effect; and a safety margin such that the drug causes no harm (Fig. 7.5). Having the correct chemical structure to confer favorable ADME properties for in vivo drug availability often involves separate structure–activity relationships than those required for primary therapeutic activity. The same is true for drug safety, i.e., eliminating any structural feature of the molecule that enables it to interact with a system in the body to cause harm (see Chapter 10). An illustration of how separate these structure–activity relationships can be is shown in Fig. 7.6. It can be seen from this figure that structural changes that have little effect on the primary activity (in this case inhibition of insulin-like growth factor receptor-1) produce a two to three order of magnitude change in a debilitating effect on cytochrome P450 enzyme inhibiton that could cause a damaging drug–drug interaction (vide infra). [1]
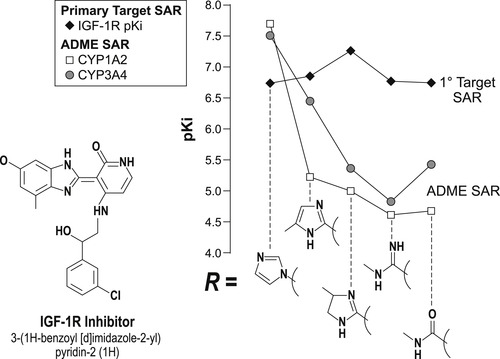 |
| Figure 7.6 Data redrawn from [1]. |
There is a general set of physico-chemical properties common to the majority of therapeutically successful drugs; these are referred to as “drug-like” properties. Drug discovery and development common experience has shown that pro-active efforts to incorporate certain drug-like properties into prospective drug candidates greatly reduce late stage attrition in the development process (see Box 7.2). It is worth considering some of the most common drug-like activities in this light.
Box 7.2
Prior to 1990, a common practice in drug discovery was to optimize primary target activity with the idea of modifying structures for ADME activity later in the process. This can be problematic since the molecular structure may be such that any changes could lead to loss of activity, i.e., there would be insufficient places in the chemical scaffold to modify ADME properties without compromising the original activity. This is illustrated by a sample of drugs shown below, where it can be seen that the original lead molecule obtained from the screening process is extraordinarily similar to the final drug. This supports the notion of having molecules in screening libraries that already have favorable ADME properties (data from [2]).
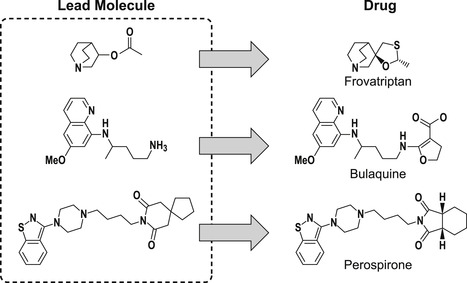 |
New Terminology
The following new terms will be introduced in this chapter:
• ADME: Acronym for absorption, distribution, metabolism and excretion, four primary elements of pharmacokinetics.
• Antedrug: Molecule that is itself an active drug but is also unstable such that as it is absorbed into the body, it is metabolized to an inactive compound.
• Bioavailability: This parameter quantifies the amount of drug that is available for physiological action in the central compartment after absorption.
• Central compartment: The main circulation (bloodstream) from where pharmacokinetic sampling of drug levels is taken.
• Clearance: The removal of drug from the body in units of volume per unit time (e.g., mL min−1).
• Dependent variable: Experimental observation taken from a system that has taken an independent variable and yielded a system response.
• Drug–drug interaction: When one drug interferes with the pharmacokinetics of another drug in the body to produce an adverse effect.
• Drug-like: The physico-chemical properties of molecules that cause them to be successful drugs in vivo.
• Enzyme induction: The reaction of the liver to pharmacokinetic stress such that levels of metabolic enzyme activity increase either through activation of gene expression, stabilization of mRNA or PXR activation.
• First pass effect: The metabolic gauntlet for drugs taken orally whereby they must first be absorbed through the gastrointestinal lumen and then immediately pass through the liver via the portal vein to be metabolized.
• Independent variable: The experimental quantity (usually drug dose or concentration) known by the experimenter that is processed by the system to yield the dependent variable.
• In vitrosystem: Latin for “within the glass,” this term refers to an experiment done in a controlled environment. For pharmacology it is when experiments are done in contained vessels and where concentration is known (a closed system).
• In vivosystem: “Within a living organism,” this term refers to experiments done in the whole body (an open system).
• Lipophilicity: The ability of a molecule to dissolve in lipids or organic solvents such as octanol or heptane, as opposed to water.
• LogP (LogD): Logarithm of the ratio of the solubility of a compound in organic medium versus aqueous medium. cLogP is calculated and mLogP is measured. Log D takes into account ionized species in the aqueous medium and must be reported with a distinct pH value.
• MAD: “Maximal absorbable dose” is the maximum amount of drug that can be expected to be absorbed from the GI tract when a compound is given orally.
• Mechanism-based inhibition (also time-dependent inhibition): Blockade of an enzyme that is essentially irreversible. The onset time for full blockade is prolonged and new enzyme activity can only be obtained through synthesis of new enzyme.
• PAMPA: “Parallel artificial membrane permeation assay” consisting of a synthetic hexadecane lipid membrane layered over a screen to allow measurement of molecules traversing the membrane via bulk lipid diffusion.
• Permeation (Papp): The rate of transfer of a molecule across a membrane (usually expressed in cm s−1).
• pKa: Negative logarithm of equilibrium equation describing relative amounts of a molecule in the acid/base form over the unionized form.
• Prodrug: Molecule that itself is not biologically active but is optimized to be absorbed into the body. Once the molecule is absorbed, biological or chemical processes transform the prodrug into the active drug.
• t½: The time required for the amount of substance, decaying as a result of an exponential process, to decrease to half of its initial value.
“Drug-Like” Properties of Molecules
The physico-chemical properties of molecules that are important to their function as drugs can be summarized under the following headings:
1. Water solubility;
2. Lipophilicity;
3. pKA and acid/base properties;
4. molecular weight.
It is worth considering these separately.
1. Water solubility: A molecule must dissolve in water before it can interact with any process in the body, including passage through lipid membranes. Molecules can have a wide range of solubility in water and this can greatly affect how well they are absorbed and metabolized, how they distribute through the body compartments and how they are excreted. An indication of how important water solubility is can be determined from consideration of MAD (maximal absorbable dose) values. This is basically the maximal amount of drug that can be absorbed orally if the small intestine (the site for oral drug absorption) were saturated with the drug (i.e., the maximum amount dissolved in water) for 4.5 hours (the transit time for intestinal contents). The MAD is given by:
(7.1)
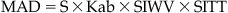
2. Lipophilicity: This is the degree to which the drug molecule will dissolve in organic media; it is a surrogate for how well the drug will dissolve in biological lipid membranes. This can be estimated through LogP or LogD values which can, in turn, be measured or calculated through indices associated with chemical groups on the molecule. LogP values are logarithms of the relative concentration of the molecule dissolved in an organic medium such as octanol or heptane versus that dissolved in water. Thus, a LogP value of 0.5 indicates a fairly water-soluble molecule with a ratio of 3.16 to 1 for octanol to water. LogP values >3 indicate highly lipid-soluble molecules (ratio octanol to water of >1000 to 1). LogD values are the same as LogP values except, unlike the latter, they also include the ionic species. For this reason they must be reported with a given pH value, as the ionization of molecules can change at different pH values (vide infra). The dependence of cellular absorption on LogP or LogD values illustrates the particular dichotomous behavior drug molecules must have toward aqueous and organic media, i.e., drugs must dissolve in water but also in lipid to interact with biological systems and cross membranes; this will be considered further in discussion of drug absorption. A drug-like value for LogP ranges from 0.5<LogP<3. LogP and Log D values are widely used in the structure–activity analyses of ADME properties, since they are readily available for molecules and can be correlated with a wide range of ADME processes. Of a sample of 1791 approved drugs, the mean LogP value is 2.5. [2]
3. Acid/base properties: Molecules can function as acids or bases in aqueous media depending on the ability of various chemical groups to lose or gain hydrogen ions. The ease with which this occurs depends on the pH of the medium. The relationship between the propensity of a molecule to be in an acidic or basic state and the pH is given by the Henderson–Hasslebach equation:
(7.2)
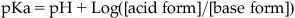
4. Molecular weight: Extremely large molecules do not cross plasma–lipid membranes well; therefore a molecular weight <350 is a good target for drug-like activity. Of a sample of all marketed drugs up to 1995, the median molecular weight is 350. However, there are classes of drug that seem to require greater mass, e.g., HIV protease and renin inhibitors have a median molecular weight of 680–700.
In general, the physico-chemical properties of a molecule should be within certain ranges for drug-like activity but exceptions to these rules can be found in every category (see Box 7.3). A frequently used guideline is the so-called “rule of 5” reported by Lipinski and colleagues. [3] and [4] From an analysis of 2245 drug-like compounds, it was observed that 89% had a molecular weight <550, only 10% had a calculated LogP >5.0, only 8% had a sum of OH and NH (hydrogen bond donors) groups >5 and only 12% had the sum of N and O atoms (hydrogen bond acceptors) >10. From these facts came a set of four rules involving the number 5 suggesting that poor absorption or permeation would be more likely from molecules that had:
Box 7.3




Guidelines for drug absorption can be derived from solubility and LogP data, but there are exceptions. For instance, azithromycin has an extremely low transintestinal absorption rate constant (Kab=0.001min−1) normally predictive of very poor absorption. However, the extraordinarily high solubility of this molecule (>50mgmL−1) allows it to have a very high MAD of 3.75g. [9]
Usually, exceedingly high LogP values are associated with lack of membrane permeation due to the fact that the molecules may lodge in the lipid membrane and not traverse the cell (i.e., dihydropyridines). High LogP values are also associated with non-specific activity and undesirable drug profiles. However, torcetrapib, a drug for dyslipidemia targeting cholesterol ester transfer protein, has a cLogP of 8.2. In this case, the natural substrates for this target are cholesterol esters (cLogP=18) thus the high LogP for torcetrapib is a requirement for activity. [10]
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
